- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 7C - Determination Of Nitrogen Oxide Emissions From Stationary Sources (Alkaline Permanganate/Colorimetric Method)
NOTE: This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 3, Method 6 and Method 7.
Content [ show/hide ].1.0 Scope and Application.
1.1 Analytes.

1.2 Applicability.
This method applies to the measurement of NOx emissions from fossil-fuel fired steam generators, electric utility plants, nitric acid plants, or other sources as specified in the regulations.
1.3 Data Quality Objectives.
Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method.
An integrated gas sample is extracted from the stack and passed through impingers containing an alkaline potassium permanganate solution; NOx (NO + NO2) emissions are oxidized to NO2- and NO3-. Then NO3- is reduced to NO2- with cadmium, and the NO2- is analyzed colorimetrically.
3.0 Definitions. [Reserved]
4.0 Interferences.
Possible interferents are sulfur dioxides (S02) and ammonia (NH3).
4.1 High concentrations of SO2 could interfere because SO2 consumes MnO4- (as does NOx) and, therefore, could reduce the NOx collection efficiency. However, when sampling emissions from a coal-fired electric utility plant burning 2.1 percent sulfur coal with no control of SO2 emissions, collection efficiency was not reduced. In fact, calculations show that sampling 3000 ppm SO2 will reduce the MnO4- concentration by only 5 percent if all the SO2 is consumed in the first impinger.
4.2 Ammonia (NH3) is slowly oxidized to NO3- by the absorbing solution. At 100 ppm NH3 in the gas stream, an interference of 6 ppm NOx (11 mg NO2/m3) was observed when the sample was analyzed 10 days after collection. Therefore, the method may not be applicable to plants using NH3 injection to control NOx emissions unless means are taken to correct the results. An equation has been developed to allow quantification of the interference and is discussed in Reference 5 of Section 16.0.
5.0 Safety.
5.1 Disclaimer.
This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents.
The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrochloric Acid (HCl).
Highly toxic and corrosive. Causes severe damage to skin. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to vapor concentrations of 0.13 to 0.2 percent can be lethal in minutes. Will react with metals, producing hydrogen.
5.2.2 Oxalic Acid (COOH)2.
Poisonous. Irritating to eyes, skin, nose, and throat.
5.2.3 Sodium Hydroxide (NaOH).
Causes severe damage to eye tissues and to skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with small amounts of water.
5.2.4 Potassium Permanganate (KMnO4).
Caustic, strong oxidizer. Avoid bodily contact with.
6.0 Equipment and Supplies.
6.1 Sample Collection and Sample Recovery.
A schematic of the Method 7C sampling train is shown in Figure 7C-1, and component parts are discussed below. Alternative apparatus and procedures are allowed provided acceptable accuracy and precision can be demonstrated to the satisfaction of the Administrator.
6.1.1 Probe. Borosilicate glass tubing, sufficiently heated to prevent water condensation and equipped with an in-stack or heated out-of-stack filter to remove particulate matter (a plug of glass wool is satisfactory for this purpose). Stainless steel or Teflon tubing may also be used for the Probe.
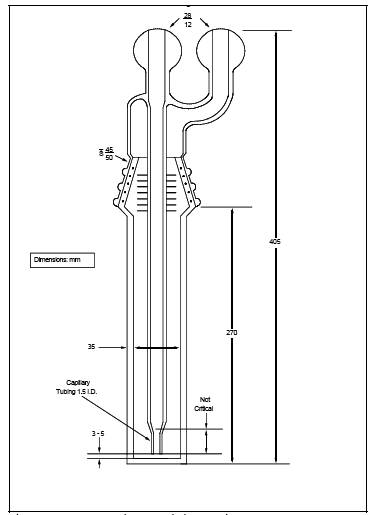
6.1.2 impingers. Three restricted-orifice glass impingers, having the specifications given in Figure 7C-2, are required for each sampling train. The impingers must be connected in series with leak-free glass connectors. Stopcock grease may be used, if necessary, to prevent leakage. (The impingers can be fabricated by a glass blower if not available commercially.)
6.1.3 glass Wool, Stopcock Grease, Drying Tube, Valve, pump, barometer, and Vacuum Gauge and Rotameter. Same as in Method 6, Sections 6.1.1.3, 6.1.1.4, 6.1.1.6, 6.1.1.7, 6.1.1.8, 6.1.2, and 6.1.3, respectively.
6.1.4 Rate meter. Rotameter, or equivalent, accurate to within 2 percent at the selected flow rate of between 400 and 500 ml/min (0.014 to 0.018 cfm). For rotameters, a range of 0 to 1 liter/min (0 to 0.035 cfm) is recommended.
6.1.5 Volume meter. console meter (DGM) capable of measuring the sample volume under the sampling conditions of 400 to 500 ml/min (0.014 to 0.018 cfm) for 60 minutes within an accuracy of 2 percent.
6.1.6 filter. To remove NOx from ambient air, prepared by adding 20 g of 5-angstrom molecular sieve to a cylindrical tube (e.g., a polyethylene drying tube).
6.1.7 Polyethylene Bottles. 1-liter, for sample recovery.
6.1.8 Funnel and Stirring Rods. For sample recovery.
6.2 Sample Preparation and Analysis.
6.2.1 Hot Plate. Stirring type with 50- by 10-mm Teflon-coated stirring bars.
6.2.2 Beakers. 400-, 600-, and 1000-ml capacities.
6.2.3 Filtering Flask. 500-ml capacity with side arm.
6.2.4 Buchner Funnel. 75-mm ID, with spout equipped with a 13-mm ID by 90-mm long piece of Teflon tubing to minimize possibility of aspirating sample solution during filtration.
6.2.5 filter Paper. Whatman GF/C, 7.0-cm diameter.
6.2.6 Stirring Rods.
6.2.7 Volumetric Flasks. 100-, 200- or 250-, 500-, and 1000-ml capacity.
6.2.8 Watch glasses. To cover 600- and 1000-ml beakers.
6.2.9 Graduated Cylinders. 50- and 250-ml capacities.
6.2.10 Pipettes. Class A.
6.2.11 pH meter. To measure pH from 0.5 to 12.0.
6.2.12 Burette. 50-ml with a micrometer type stopcock. (The stopcock is Catalog No. 8225-t-05, Ace glass, Inc., Post Office Box 996, Louisville, Kentucky 50201.) Place a glass wool plug in bottom of burette. Cut off burette at a height of 43 cm (17 in.) from the top of plug, and have a blower attach a glass funnel to top of burette such that the diameter of the burette remains essentially unchanged. Other means of attaching the funnel are acceptable.
6.2.13 glass Funnel. 75-mm ID at the top.
6.2.14 Spectrophotometer. Capable of measuring absorbance at 540 nm; 1-cm cells are adequate.
6.2.15 Metal Thermometers. Bimetallic thermometers, range 0 to 150 C (32 to 300 F).
6.2.16 Culture Tubes. 20- by 150-mm, Kimax No. 45048.
6.2.17 Parafilm "M." Obtained from American Can Company, Greenwich, Connecticut 06830.
6.2.18 CO2 Measurement equipment. Same as in Method 3, Section 6.0.
7.0 Reagents and Standards.
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection.
7.1.1 Water. Deionized distilled to conform to ASTM Specification D 1193-77 or 91 Type 3 (incorporated by reference - see '60.17).
7.1.2 Potassium Permanganate, 4.0 Percent (w/w), Sodium Hydroxide, 2.0 Percent (w/w) solution (KMnO4/NaOH solution). Dissolve 40.0 g of KMnO4 and 20.0 g of NaOH in 940 ml of water.
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in Section 7.1.1.
7.2.2 Oxalic Acid Solution. Dissolve 48 g of oxalic acid [(COOH)2 2H2O] in water, and dilute to 500 ml. Do not heat the solution.
7.2.3 Sodium Hydroxide, 0.5 N. Dissolve 20 g of NaOH in water, and dilute to 1 liter.
7.2.4 Sodium Hydroxide, 10 N. Dissolve 40 g of NaOH in water, and dilute to 100 ml.
7.2.5 Ethylenediamine Tetraacetic Acid (EDTA) Solution, 6.5 percent (w/v). Dissolve 6.5 g of EDTA (disodium salt) in water, and dilute to 100 ml. Dissolution is best accomplished by using a magnetic stirrer.
7.2.6 Column Rinse Solution. Add 20 ml of 6.5 percent EDTA solution to 960 ml of water, and adjust the pH to between 11.7 and 12.0 with 0.5 N NaOH.
7.2.7 Hydrochloric Acid (HCl), 2 N. Add 86 ml of concentrated HCl to a 500 ml-volumetric flask containing water, dilute to volume, and mix well. Store in a glass-stoppered bottle.
7.2.8 Sulfanilamide Solution. Add 20 g of sulfanilamide [melting point 165 to 167 C (329 to 333 F)] to 700 ml of water. Add, with mixing, 50 ml concentrated phosphoric acid (85 percent), and dilute to 1000 ml. This solution is stable for at least 1 month, if refrigerated.
7.2.9 N-(1-Naphthyl)-Ethylenediamine Dihydrochloride (NEDA) Solution. Dissolve 0.5 g of NEDA in 500 ml of water. An aqueous solution should have one absorption peak at 320 nm over the range of 260 to 400 nm. NEDA that shows more than one absorption peak over this range is impure and should not be used. This solution is stable for at least 1 month if protected from light and refrigerated.
7.2.10 Cadmium. Obtained from Matheson Coleman and Bell, 2909 Highland Avenue, Norwood, Ohio 45212, as EM Laboratories Catalog No. 2001. Prepare by rinsing in 2 N HCl for 5 minutes until the color is silver-grey. Then rinse the cadmium with water until the rinsings are neutral when tested with pH paper. CAUTION: H2 is liberated during preparation. Prepare in an exhaust hood away from any flame or combustion source.
7.2.11 Sodium Sulfite (NaNO2) Standard Solution, Nominal Concentration, 1000 g NO2-/ml. Desiccate NaNO2 overnight. Accurately weigh 1.4 to 1.6 g of NaNO2 (assay of 97 percent NaNO2 or greater), dissolve in water, and dilute to 1 liter. Calculate the exact NO2- concentration using Equation 7C-1 in Section 12.2. This solution is stable for at least 6 months under laboratory conditions.
7.2.12 Potassium Nitrate (KNO3) Standard Solution. Dry KNO3 at 110 C (230 F) for 2 hours, and cool in a desiccator. Accurately weigh 9 to 10 g of KNO3 to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact NO3- concentration using Equation 7C-2 in Section 12.3. This solution is stable for 2 months without preservative under laboratory conditions.
7.2.13 Spiking Solution. Pipette 7 ml of the KNO3 standard into a 100-ml volumetric flask, and dilute to volume.
7.2.14 Blank Solution. Dissolve 2.4 g of KMnO4 and 1.2 g of NaOH in 96 ml of water. Alternatively, dilute 60 ml of KMnO4/NaOH solution to 100 ml.
7.2.15 Quality Assurance Audit Samples. Same as in Method 7, Section 7.3.10. When requesting audit samples, specify that they be in the appropriate concentration range for Method 7C.
8.0 Sample Collection, Preservation, Storage, and Transport.
8.1 Preparation of Sampling Train.
Add 200 ml of KMnO4/NaOH solution (Section 7.1.2) to each of three impingers, and assemble the train as shown in Figure 7C-1. Adjust the Probe heater to a temperature sufficient to prevent water condensation.
8.2 Leak-Checks.
Same as in Method 6, Section 8.2.
8.3 Sample Collection.
8.3.1 Record the initial DGM reading and barometric pressure. Determine the sampling point or points according to the appropriate regulations (e.g., ' 60.46(b)(5) of 40 CFR Part 60). Position the tip of the Probe at the sampling point, connect the Probe to the first impinger, and start the pump. Adjust the sample flow to a value between 400 and 500 ml/min (0.014 and 0.018 cfm). CAUTION: DO NOT EXCEED THESE flow RATES. Once adjusted, maintain a constant flow rate during the entire sampling run. Sample for 60 minutes. For relative accuracy (RA) testing of continuous emission monitors, the minimum sampling time is 1 hour, sampling 20 minutes at each traverse point.
NOTE: When the SO2 concentration is greater than 1200 ppm, the sampling time may have to be reduced to 30 minutes to eliminate plugging of the impinger orifice with MnO2. For RA tests with SO2 greater than 1200 ppm, sample for 30 minutes (10 minutes at each point).
8.3.2 Record the DGM temperature, and check the flow rate at least every 5 minutes. At the conclusion of each run, turn off the pump, remove the Probe from the stack, and record the final readings. Divide the sample volume by the sampling time to determine the average flow rate. Conduct the mandatory post-test leak-check. If a leak is found, void the test run, or use procedures acceptable to the Administrator to adjust the sample volume for the leakage.
8.4 CO2 Measurement.
During sampling, measure the CO2 content of the stack gas near the sampling point using Method 3. The single-point grab sampling procedure is adequate, provided the measurements are made at least three times (near the start, midway, and before the end of a run), and the average CO2 concentration is computed. The Orsat or Fyrite analyzer may be used for this analysis.
8.5 Sample Recovery.
Disconnect the impingers. Pour the contents of the impingers into a 1-liter polyethylene bottle using a funnel and a stirring rod (or other means) to prevent spillage. Complete the quantitative transfer by rinsing the impingers and connecting tubes with water until the rinsings are clear to light pink, and add the rinsings to the bottle. Mix the sample, and mark the solution level. Seal and identify the sample container.
9.0 Quality Control.
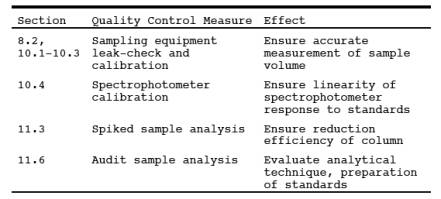
10.0 Calibration and Standardizations.
10.1 Volume metering System. Same as Method 6, Section 10.1. For detailed instructions on carrying out these calibrations, it is suggested that Section 3.5.2 of Reference 4 of Section 16.0 be consulted.
10.2 tenperature sensors and barometer. Same as in Method 6, Sections 10.2 and 10.4, respectively.
10.3 Check of Rate meter calibration Accuracy (Optional). Disconnect the Probe from the first impinger, and connect the filter. Start the pump, and adjust the rate meter to read between 400 and 500 ml/min (0.014 and 0.018 cfm). After the flow rate has stabilized, start measuring the volume sampled, as recorded by the console meter and the sampling time. Collect enough volume to measure accurately the flow rate. Then calculate the flow rate. This average flow rate must be less than 500 ml/min (0.018 cfm) for the sample to be valid; therefore, it is recommended that the flow rate be checked as above prior to each test.
10.4 Spectrophotometer.
10.4.1 Dilute 5.0 ml of the NaNO2 standard solution to 200 ml with water. This solution nominally contains 25 g NO2 -/ml. Use this solution to prepare calibration standards to cover the range of 0.25 to 3.00 g NO2-/ml. Prepare a minimum of three standards each for the linear and slightly nonlinear (described below) range of the curve. Use pipettes for all additions.
10.4.2 Measure the absorbance of the standards and a water blank as instructed in Section 11.5. Plot the net absorbance vs. g NO2-/ml. Draw a smooth curve through the points. The curve should be linear up to an absorbance of approximately 1.2 with a slope of approximately 0.53 absorbance units/g NO2-/ml. The curve should pass through the origin. The curve is slightly nonlinear from an absorbance of 1.2 to 1.6.
11.0 Analytical Procedures.
11.1 Sample Stability.
Collected samples are stable for at least four weeks; thus, analysis must occur within 4 weeks of collection.
11.2 Sample Preparation.
11.2.1 Prepare a cadmium reduction column as follows: Fill the burette with water. Add freshly prepared cadmium slowly, with tapping, until no further settling occurs. The height of the cadmium column should be 39 cm (15 in). When not in use, store the column under rinse solution.
NOTE: The column should not contain any bands of cadmium fines. This may occur if regenerated cadmium is used and will greatly reduce the column lifetime.
11.2.2 Note the level of liquid in the sample container, and determine whether any sample was lost during shipment. If a noticeable amount of leakage has occurred, the volume lost can be determined from the difference between initial and final solution levels, and this value can then be used to correct the analytical result. Quantitatively transfer the contents to a 1-liter volumetric flask, and dilute to volume.
11.2.3 Take a 100-ml aliquot of the sample and blank (unexposed KMnO4/NaOH) solutions, and transfer to 400-ml beakers containing magnetic stirring bars. Using a pH meter, add concentrated H2SO4 with stirring until a pH of 0.7 is obtained. Allow the solutions to stand for 15 minutes. Cover the beakers with watch glasses, and bring the temperature of the solutions to 50 C (122 F). Keep the temperature below 60 C (140 F). Dissolve 4.8 g of oxalic acid in a minimum volume of water, approximately 50 ml, at room temperature. Do not heat the solution. Add this solution slowly, in increments, until the KMnO4 solution becomes colorless. If the color is not completely removed, prepare some more of the above oxalic acid solution, and add until a colorless solution is obtained. Add an excess of oxalic acid by dissolving 1.6 g of oxalic acid in 50 ml of water, and add 6 ml of this solution to the colorless solution. If suspended matter is present, add concentrated H2SO4 until a clear solution is obtained.
11.2.4 Allow the samples to cool to near room temperature, being sure that the samples are still clear. Adjust the pH to between 11.7 and 12.0 with 10 N NaOH. Quantitatively transfer the mixture to a Buchner funnel containing GF/C filter paper, and filter the precipitate. filter the mixture into a 500-ml filtering flask. Wash the solid material four times with water. When filtration is complete, wash the Teflon tubing, quantitatively transfer the filtrate to a 500-ml volumetric flask, and dilute to volume. The samples are now ready for cadmium reduction. Pipette a 50-ml aliquot of the sample into a 150-ml beaker, and add a magnetic stirring bar. Pipette in 1.0 ml of 6.5 percent EDTA solution, and mix.
11.3 Determine the correct stopcock setting to establish a flow rate of 7 to 9 ml/min of column rinse solution through the cadmium reduction column. Use a 50-ml graduated cylinder to collect and measure the solution volume. After the last of the rinse solution has passed from the funnel into the burette, but before air entrapment can occur, start adding the sample, and collect it in a 250-ml graduated cylinder. Complete the quantitative transfer of the sample to the column as the sample passes through the column. After the last of the sample has passed from the funnel into the burette, start adding 60 ml of column rinse solution, and collect the rinse solution until the solution just disappears from the funnel. Quantitatively transfer the sample to a 200-ml volumetric flask (a 250-ml flask may be required), and dilute to volume. The samples are now ready for NO2- analysis.
NOTE: Two spiked samples should be run with every group of samples passed through the column. To do this, prepare two additional 50-ml aliquots of the sample suspected to have the highest NO>2- concentration, and add 1 ml of the spiking solution to these aliquots. If the spike recovery or column efficiency (see Section 12.2) is below 95 percent, prepare a new column, and repeat the cadmium reduction.
11.4 Repeat the procedures outlined in Sections 11.2 and 11.3 for each sample and each blank.
11.5 Sample Analysis. Pipette 10 ml of sample into a culture tube. Pipette in 10 ml of sulfanilamide solution and 1.4 ml of NEDA solution. Cover the culture tube with parafilm, and mix the solution. Prepare a blank in the same manner using the sample from treatment of the unexposed KMnO4/NaOH solution. Also, prepare a calibration standard to check the slope of the calibration curve. After a 10-minute color development interval, measure the absorbance at 540 nm against water. Read g NO2-/ml from the calibration curve. If the absorbance is greater than that of the highest calibration standard, use less than 10 ml of sample, and repeat the analysis. Determine the NO2- concentration using the calibration curve obtained in Section 10.4.
NOTE: Some test tubes give a high blank NO>2- value but culture tubes do not.
11.6 Audit Sample Analysis. Same as in Method 7, Section 11.4.
12.0 Data Analysis and Calculations.
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature.
| B | = | Analysis of blank, g NO2-/ml. |
| C | = | Concentration of NOx as NO2, dry basis, mg/dsm3. |
| E | = | Column efficiency, dimensionless |
| K2 | = | 10-3 mg/g. |
| m | = | Mass of NOx, as NO2, in sample, g. |
| Pbar | = | Barometric pressure, mm Hg (in. Hg). |
| Pstd | = | Standard absolute pressure, 760 mm Hg (29.92 in. Hg). |
| s | = | Concentration of spiking solution, g NO3/ml. |
| S | = | Analysis of sample, g NO2-/ml. |
| Tm | = | Average console meter absolute temperature, K. |
| Tstd | = | Standard absolute temperature, 293 K (528 R). |
| Vm(std) | = | Dry gas volume measured by the console meter, corrected to standard conditions, dscm (dscf). |
| Vm | = | Dry gas volume as measured by the console meter, scm (scf). |
| x | = | Analysis of spiked sample, g NO2-/ml. |
| X | = | Correction factor for CO2 collection |
| = | 100/[100 - %CO2(V/V)]. | |
| y | = | Analysis of unspiked sample, g NO2-/ml. |
| Y | = | console meter calibration factor. |
| 1.0 ppm NO | = | 1.247 mg NO/m3 at STP. |
| 1.0 ppm NO2 | = | 1.912 mg NO2/m3 at STP. |
| 1 ft3 | = | 2.832 x 10-2 m3. |
12.2 NO2 Concentration. Calculate the NO2concentration of the solution (see Section 7.2.11) using the following equation:
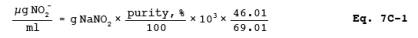
12.3 NO3 Concentration. Calculate the NO3 concentration of the KNO3 solution (see Section 7.2.12) using the following equation:
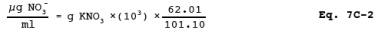
12.4 Sample Volume, Dry Basis, Corrected to Standard Conditions.

where:
| K1 | = | 0.3855 K/mm Hg for metric units. |
| = | 17.65 R/in. Hg for English units. |
12.5 Efficiency of Cadmium Reduction Column. Calculate this value as follows:
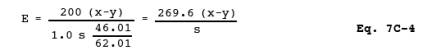
where:
| 200 | = | Final volume of sample and blank after passing through the column, ml. |
| 1.0 | = | Volume of spiking solution added, ml. |
| 46.01 | = | g NO2-/mole. |
| 62.01 | = | g NO3-/mole. |
12.6 Total g NO2.
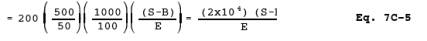
where:
| 500 | = | Total volume of prepared sample, ml. |
| 50 | = | Aliquot of prepared sample processed through cadmium column, ml. |
| 100 | = | Aliquot of KMnO4/NaOH solution, ml. |
| 1000 | = | Total volume of KMnO4/NaOH solution, ml. |
12.7 Sample Concentration.

13.0 Method Performance.
13.1 Precision. The intra-laboratory relative standard deviation for a single measurement is 2.8 and 2.9 percent at 201 and 268 ppm NOx, respectively.
13.2 Bias. The method does not exhibit any bias relative to Method 7.
13.3 Range. The lower detectable limit is 13 mg NOx/m3x).
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
1. Margeson, J.H., W.J. Mitchell, J.C. Suggs, and M.R. Midgett. Integrated Sampling and Analysis Methods for Determining NOx Emissions at Electric Utility Plants. U.S. Environmental Protection Agency, Research Triangle Park, NC. Journal of the Air Pollution Control Association. 32:1210-1215. 1982.
2. Memorandum and attachment from J.H. Margeson, Source Branch, Quality Assurance Division, Environmental Monitoring Systems Laboratory, to The Record, EPA. March 30, 1983. NH3 Interference in Methods 7C and 7D.
3. Margeson, J.H., J.C. Suggs, and M.R. Midgett. Reduction of Nitrate to Nitrite with Cadmium. Anal. Chem. 52:1955-57. 1980.
4. Quality Assurance Handbook for Air Pollution Measurement Systems. Volume III - Stationary Source Specific Methods. U.S. Environmental Protection Agency. Research Triangle Park, NC. Publication No. EPA-600/4-77-027b. August 1977.
5. Margeson, J.H., et al. An Integrated Method for Determining NOx Emissions at Nitric Acid Plants. Analytical Chemistry. 47 (11):1801. 1975.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
Figure 7C-1. NOx Sampling Train.
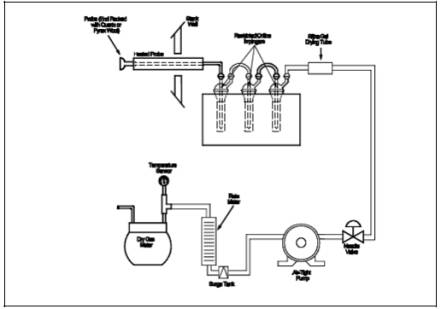
Figure 7C-2. Restricted-Orifice impinger.
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources