- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 7 - Determination Of Nitrogen Oxide Emissions From Stationary Sources
NOTE: This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1 and Method 5.
Content [ show/hide ].1.0 Scope and Application.
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
| Nitrogen oxides(NOx), as NO2, including: Nitric oxide (NO) Nitrogen dioxide (NO2) |
10102 - 43 - 9 10102 - 44 - 0 |
2 - 400 mg/dscm |
1.2 Applicability.
This method is applicable for the measurement of nitrogen oxides (NOx) emitted from stationary sources.
1.3 Data Quality Objectives.
Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sample methods.
2.0 Summary of Method.
A grab sample is collected in an evacuated flask containing a dilute sulfuric acid-hydrogen peroxide absorbing solution, and the nitrogen oxides, except nitrous oxide, are measured colorimetrically using the phenoldisulfonic acid (PDS) procedure.
3.0 Definitions. [Reserved]
4.0 Interferences.
Biased results have been observed when sampling under conditions of high sulfur dioxide concentrations (above 2000 ppm).
5.0 Safety.
5.1 Disclaimer.
This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents.
The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2).
Irritating to eyes, skin, nose, and lungs.
5.2.2 Phenoldisulfonic Acid.
Irritating to eyes and skin.
5.2.3 Sodium Hydroxide (NaOH).
Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.4 Sulfuric Acid (H2SO4).
Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
5.2.5 Phenol.
Poisonous and caustic. Do not handle with bare hands as it is absorbed through the skin.
6.0 Equipment and Supplies.
6.1 Sample Collection.
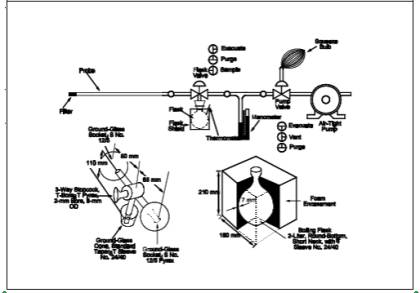
A schematic of the sampling train used in performing this method is shown in Figure 7-1. Other grab sampling systems or equipment, capable of measuring sample volume to within 2.0 percent and collecting a sufficient sample volume to allow analytical reproducibility to within 5 percent, will be considered acceptable alternatives, subject to the approval of the Administrator. The following items are required for sample collection:
6.1.1 Probe.
Borosilicate glass tubing, sufficiently heated to prevent water condensation and equipped with an in-stack or heated out-of-stack filter to remove particulate matter (a plug of glass wool is satisfactory for this purpose). Stainless steel or Teflon tubing may also be used for the Probe. Heating is not necessary if the Probe remains dry during the purging period.
6.1.2 Collection Flask.
Two-liter borosilicate, round bottom flask, with short neck and 24/40 standard taper opening, protected against implosion or breakage.
6.1.3 Flask Valve.
T-bore stopcock connected to a 24/40 standard taper joint.
6.1.4 temperature Gauge.
Dial-type thermometer, or other temperature gauge, capable of measuring 1 C (2 F) intervals from -5 to 50 C (23 to 122 F).
6.1.5 Vacuum Line.
tubing capable of withstanding a vacuum of 75 mm (3 in.) Hg absolute pressure, with "T" connection and T-bore stopcock.
6.1.6 Vacuum Gauge.
U-tube manometer, 1 meter (39 in.), with 1 mm (0.04 in.) divisions, or other gauge capable of measuring pressure to within 2.5 mm (0.10 in.) Hg.
6.1.7 pump.
Capable of evacuating the collection flask to a pressure equal to or less than 75 mm (3 in.) Hg absolute.
6.1.8 Squeeze Bulb.
One-way.
6.1.9 Volumetric Pipette. 25-ml.
6.1.10 Stopcock and Ground Joint Grease.
A highvacuum, high-temperature chlorofluorocarbon grease is required. Halocarbon 25-5S has been found to be effective.
6.1.11 barometer.
Mercury, aneroid, or other barometer capable of measuring atmospheric pressure to within 2.5 mm (0.1 in.) Hg. See NOTE in Method 5, Section 6.1.2.
6.2 Sample Recovery.
The following items are required for sample recovery:
6.2.1 Graduated Cylinder.
50-ml with 1 ml divisions.
6.2.2 Storage Containers.
Leak-free polyethylene bottles.
6.2.3 Wash Bottle.
Polyethylene or glass.
6.2.4 glass Stirring Rod.
6.2.5 Test Paper for Indicating pH.
To cover the pH range of 7 to 14.
6.3 Analysis.
The following items are required for analysis:
6.3.1 Volumetric Pipettes.
Two 1-ml, two 2-ml, one 3-ml, one 4-ml, two 10-ml, and one 25-ml for each sample and standard.
6.3.2 Porcelain Evaporating Dishes.
175- to 250-ml capacity with lip for pouring, one for each sample and each standard. The Coors No. 45006 (shallowform, 195-ml) has been found to be satisfactory. Alternatively, polymethyl pentene beakers (Nalge No. 1203, 150-ml), or glass beakers (150-ml) may be used. When glass beakers are used, etching of the beakers may cause solid matter to be present in the analytical step; the solids should be removed by filtration.
6.3.3 Steam Bath.
Low-temperature ovens or thermostatically controlled hot plates kept below 70 C (160 F) are acceptable alternatives.
6.3.4 Dropping Pipette or Dropper.
Three required.
6.3.5 Polyethylene Policeman.
One for each sample and each standard.
6.3.6 Graduated Cylinder.
100-ml with 1-ml divisions.
6.3.7 Volumetric Flasks.
50-ml (one for each sample and each standard), 100-ml (one for each sample and each standard, and one for the working standard KNO3solution), and 1000-ml (one).
6.3.8 Spectrophotometer.
To measure at 410 nm.
6.3.9 Graduated Pipette.
10-ml with 0.1-ml divisions.
6.3.10 Test Paper for Indicating pH.
To cover the pH range of 7 to 14.
6.3.11 Analytical Balance.
To measure to within 0.1 mg.
7.0 Reagents and Standards.
Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection.
The following reagents are required for sampling:
7.1.1 Water.
Deionized distilled to conform to ASTM D 1193-77 or 91 Type 3 (incorporated by reference - see 60.17). The KMnO4 test for oxidizable organic matter may be omitted when high concentrations of organic matter are not expected to be present.
7.1.2 Absorbing Solution.
Cautiously add 2.8 ml concentrated H2SO4 to a 1-liter flask partially filled with water. Mix well, and add 6 ml of 3 percent hydrogen peroxide, freshly prepared from 30 percent hydrogen peroxide solution. Dilute to 1 liter of water and mix well. The absorbing solution should be used within 1 week of its preparation. Do not expose to extreme heat or direct sunlight.
7.2 Sample Recovery.
The following reagents are required for sample recovery:
7.2.1 Water.
Same as in 7.1.1.
7.2.2 Sodium Hydroxide
1 N. Dissolve 40 g NaOH in water, and dilute to 1 liter.
7.3 Analysis.
The following reagents and standards are required for analysis:
7.3.1 Water.
Same as in 7.1.1.
7.3.2 Fuming Sulfuric Acid.
15 to 18 percent by weight free sulfur trioxide. HANDLE WITH CAUTION.
7.3.3 Phenol.
White solid.
7.3.4 Sulfuric Acid.
Concentrated, 95 percent minimum assay.
7.3.5 Potassium Nitrate (KNO3).
Dried at 105 to 110 C (221 to 230 F) for a minimum of 2 hours just prior to preparation of standard solution.
7.3.6 Standard KNO3 Solution.
Dissolve exactly 2.198 g of dried KNO3 in water, and dilute to 1 liter with water in a 1000-ml volumetric flask.
7.3.7 Working Standard KNO3 Solution.
Dilute 10 ml of the standard solution to 100 ml with water. One ml of the working standard solution is equivalent to 100 g nitrogen dioxide (NO2).
7.3.8 Phenoldisulfonic Acid Solution.
Dissolve 25 g of pure white phenol solid in 150 ml concentrated sulfuric acid on a steam bath. Cool, add 75 ml fuming sulfuric acid (15 to 18 percent by weight free sulfur trioxide - HANDLE WITH CAUTION), and heat at 100 C (212 F) for 2 hours. Store in a dark, stoppered bottle.
7.3.9 Concentrated Ammonium Hydroxide.
7.3.10 Quality Assurance Audit Samples.
When making compliance determinations, and upon availability, audit samples may be obtained from the appropriate EPA Regional Office or from the responsible enforcement authority. NOTE: The responsible enforcement authority should be notified at least 30 days prior to the test date to allow sufficient time for sample delivery.
8.0 Sample Collection, Preservation, Storage and Transport.
8.1 Sample Collection.
8.1.1 Flask Volume.
The volume of the collection flask and flask valve combination must be known prior to sampling. Assemble the flask and flask valve, and fill with water to the stopcock. Measure the volume of water to ± 10 ml. Record this volume on the flask.
8.1.2 Pipette 25 ml of absorbing solution into a sample flask, retaining a sufficient quantity for use in preparing the calibration standards. Insert the flask valve stopper into the flask with the valve in the "purge" position. Assemble the sampling train as shown in Figure 7-1, and place the Probe at the sampling point. Make sure that all fittings are tight and leak-free, and that all ground glass joints have been greased properly with a high-vacuum, high- temperature chlorofluorocarbon-based stopcock grease. Turn the flask valve and the pump valve to their "evacuate" positions. Evacuate the flask to 75 mm (3 in.) Hg absolute pressure, or less. Evacuation to a pressure approaching the vapor pressure of water at the existing temperature is desirable. Turn the pump valve to its "vent" position, and turn off the pump. Check for leakage by observing the manometer for any pressure fluctuation. (Any variation greater than 10 mm (0.4 in.) Hg over a period of 1 minute is not acceptable, and the flask is not to be used until the leakage problem is corrected. Pressure in the flask is not to exceed 75 mm (3 in.) Hg absolute at the time sampling is commenced.) Record the volume of the flask and valve (Vf), the flask temperature (Ti), and the barometric pressure. Turn the flask valve counterclockwise to its "purge" position, and do the same with the pump valve. Purge the Probe and the vacuum tube using the squeeze bulb. If condensation occurs in the Probe and the flask valve area, heat the Probe, and purge until the condensation disappears. Next, turn the pump valve to its "vent" position. Turn the flask valve clockwise to its "evacuate" position, and record the difference in the mercury levels in the manometer. The absolute internal pressure in the flask (Pi) is equal to the barometric pressure less the manometer reading. Immediately turn the flask valve to the "sample" position, and permit the gas to enter the flask until pressures in the flask and sample line (i.e., duct, stack) are equal. This will usually require about 15 seconds; a longer period indicates a plug in the Probe, which must be corrected before sampling is continued. After collecting the sample, turn the flask valve to its "purge" position, and disconnect the flask from the sampling train.
8.1.3 Shake the flask for at least 5 minutes.
8.1.4 If the gas being sampled contains insufficient oxygen for the conversion of NO to NO2 (e.g., an applicable subpart of the standards may require taking a sample of a calibration gas mixture of NO in N2), then introduce oxygen into the flask to permit this conversion. Oxygen may be introduced into the flask by one of three methods: (1) Before evacuating the sampling flask, flush with pure cylinder oxygen, then evacuate flask to 75 mm (3 in.) Hg absolute pressure or less; or (2) inject oxygen into the flask after sampling; or (3) terminate sampling with a minimum of 50 mm (2 in.) Hg vacuum remaining in the flask, record this final pressure, and then vent the flask to the atmosphere until the flask pressure is almost equal to atmospheric pressure.
8.2 Sample Recovery.
Let the flask sit for a minimum of 16 hours, and then shake the contents for 2 minutes.
8.2.1 Connect the flask to a mercury filled U-tube manometer. Open the valve from the flask to the manometer, and record the flask temperature (Tf), the barometric pressure, and the difference between the mercury levels in the manometer. The absolute internal pressure in the flask (Pf) is the barometric pressure less the manometer reading. Transfer the contents of the flask to a leak-free polyethylene bottle. Rinse the flask twice with 5 ml portions of water, and add the rinse water to the bottle. Adjust the pH to between 9 and 12 by adding 1 N NaOH, dropwise (about 25 to 35 drops). Check the pH by dipping a stirring rod into the solution and then touching the rod to the pH test paper. Remove as little material as possible during this step. Mark the height of the liquid level so that the container can be checked for leakage after transport. Label the container to identify clearly its contents. Seal the container for shipping.
9.0 Quality Control.
| Section | Quality Control Measure | Effect |
| 10.1 | Spectrophometer calibration | Ensure linearity of spectrophometer to respond to standards |
| 11.4 | Audit sample analysis | Evaluate analytical technique, preparation of standard |
10.0 Calibration and Standardization.
10.1 Spectrophotometer.
10.1.1 Optimum Wavelength Determination.
10.1.1.1 Calibrate the wavelength scale of the spectrophotometer every 6 months. The calibration may be accomplished by using an energy source with an intense line emission such as a mercury lamp, or by using a series of glass filters spanning the measuring range of the spectrophotometer. calibration materials are available commercially and from the National Institute of Standards and Technology. Specific details on the use of such materials should be supplied by the vendor; general information about calibration techniques can be obtained from general reference books on analytical chemistry. The wavelength scale of the spectrophotometer must read correctly within 5 nm at all calibration points; otherwise, repair and recalibrate the spectrophotometer. Once the wavelength scale of the spectrophotometer is in proper calibration, use 410 nm as the optimum wavelength for the measurement of the absorbance of the standards and samples.
10.1.1.2 Alternatively, a scanning procedure may be employed to determine the proper measuring wavelength. If the instrument is a double-beam spectrophotometer, scan the spectrum between 400 and 415 nm using a 200 g NO2 standard solution in the sample cell and a blank solution in the reference cell. If a peak does not occur, the spectrophotometer is probably malfunctioning and should be repaired. When a peak is obtained within the 400 to 415 nm range, the wavelength at which this peak occurs shall be the optimum wavelength for the measurement of absorbance of both the standards and the samples. For a single-beam spectrophotometer, follow the scanning procedure described above, except scan separately the blank and standard solutions. The optimum wavelength shall be the wavelength at which the maximum difference in absorbance between the standard and the blank occurs.
10.1.2 Determination of Spectrophotometer calibration Factor Kc.
to a series of five 50-ml volumetric flasks. To each flask, add 25 ml of absorbing solution and 10 ml water. Add 1 N NaOH to each flask until the pH is between 9 and 12 (about 25 to 35 drops). Dilute to the mark with water. Mix thoroughly, and pipette a 25-ml aliquot of each solution into a separate porcelain-evaporating dish. Beginning with the evaporation step, follow the analysis procedure of Section 11.2 until the solution has been transferred to the 100-ml volumetric flask and diluted to the mark. Measure the absorbance of each solution at the optimum wavelength as determined in Section 10.2.1. This calibration procedure must be repeated on each day that samples are analyzed. Calculate the spectrophotometer calibration factor as shown in Section 12.2.
10.1.3 Spectrophotometer calibration Quality Control.
Multiply the absorbance value obtained for each standard by the Kc factor (reciprocal of the least squares slope) to determine the distance each calibration point lies from the theoretical calibration line. The difference between the calculated concentration values and the actual concentrations (i.e., 100, 200, 300, and 400 μg NO2) should be less than 7 percent for all standards.
10.2 barometer.
Calibrate against a mercury barometer.
10.3 temperature Gauge.
Calibrate dial thermometers against mercury-in-glass thermometers.
10.4 Vacuum Gauge.
Calibrate mechanical gauges, if used, against a mercury manometer such as that specified in Section 6.1.6.
10.5 Analytical Balance.
Calibrate against standard weights.
11.0 Analytical Procedures.
11.1 Sample Loss Check.
Note the level of the liquid in the container, and confirm whether any sample was lost during shipment. Note this on the analytical data sheet. If a noticeable amount of leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.2 Sample Preparation.
Immediately prior to analysis, transfer the contents of the shipping container to a 50 ml volumetric flask, and rinse the container twice with 5 ml portions of water. Add the rinse water to the flask, and dilute to mark with water; mix thoroughly. Pipette a 25-ml aliquot into the porcelain evaporating dish. Return any unused portion of the sample to the polyethylene storage bottle. Evaporate the 25-ml aliquot to dryness on a steam bath, and allow to cool. Add 2 ml phenoldisulfonic acid solution to the dried residue, and triturate thoroughly with a polyethylene policeman. Make sure the solution contacts all the residue. Add 1 ml water and 4 drops of concentrated sulfuric acid. Heat the solution on a steam bath for 3 minutes with occasional stirring. Allow the solution to cool, add 20 ml water, mix well by stirring, and add concentrated ammonium hydroxide, dropwise, with constant stirring, until the pH is 10 (as determined by pH paper). If the sample contains solids, these must be removed by filtration (centrifugation is an acceptable alternative, subject to the approval of the Administrator) as follows: Filter through Whatman No. 41 filter paper into a 100-ml volumetric flask. Rinse the evaporating dish with three 5- ml portions of water. Filter these three rinses. Wash the filter with at least three 15-ml portions of water. Add the filter washings to the contents of the volumetric flask, and dilute to the mark with water. If solids are absent, the solution can be transferred directly to the 100-ml volumetric flask and diluted to the mark with water.
11.3 Sample Analysis.
Mix the contents of the flask thoroughly, and measure the absorbance at the optimum wavelength used for the standards (Section 10.2.1), using the blank solution as a zero reference. Dilute the sample and the blank with equal volumes of water if the absorbance exceeds A4, the absorbance of the 400-g NO2 standard (see Section 10.2.2).
11.4 Audit Sample Analysis.
11.4.1 When the method is used to analyze samples to demonstrate compliance with a source emission regulation, an audit sample must be analyzed, subject to availability.
11.4.2 Concurrently analyze the audit sample and the compliance samples in the same manner to evaluate the technique of the analyst and the standards preparation.
11.4.3 The same analyst, analytical reagents, and analytical system must be used for the compliance samples and the audit sample. If this condition is met, duplicate auditing of subsequent compliance analyses for the same enforcement agency within a 30-day period is waived. An audit sample set may not be used to validate different sets of compliance samples under the jurisdiction of separate enforcement agencies, unless prior arrangements have been made with both enforcement agencies.
11.5 Audit Sample Results.
11.5.1 Calculate the audit sample concentrations and submit results using the instructions provided with the audit samples.
11.5.2 Report the results of the audit samples and the compliance determination samples along with their identification numbers, and the analyst's name to the responsible enforcement authority. Include this information with reports of any subsequent compliance analyses for the same enforcement authority during the 30-day period.
11.5.3 The concentrations of the audit samples obtained by the analyst must agree within 5 percent of the actual concentration. If the 5 percent specification is not met, reanalyze the compliance and audit samples, and include initial and reanalysis values in the test report.
11.5.4 Failure to meet the 5-percent specification may require retests until the audit problems are resolved. However, if the audit results do not affect the compliance or noncompliance status of the affected facility, the Administrator may waive the reanalysis requirement, further audits, or retests and accept the results of the compliance test. While steps are being taken to resolve audit analysis problems, the Administrator may also choose to use the data to determine the compliance or noncompliance status of the affected facility.
12.0 Data Analysis and Calculations.
Carry out the calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculations.
12.1 Nomenclature.
| A | = | Absorbance of sample. |
| A1 | = | Absorbance of the 100-g NO2 standard. |
| A2 | = | Absorbance of the 200-g NO2 standard. |
| A3 | = | Absorbance of the 300-g NO2 standard. |
| A4 | = | Absorbance of the 400-g NO2 standard. |
| C | = | Concentration of NOx as NO3 (lb/dscf). |
| Cd | = | Determined audit sample concentration, mg/dscm. |
| Ca | = | Actual audit sample concentration, mg/dscm. |
| F | = | Dilution factor (i.e., 25/5, 25/10, etc., required only if sample dilution was needed to reduce the absorbance into the range of the calibration). |
| Kc | = | Spectrophotometer calibration factor. |
| m | = | Mass of NOx as NO2 in gas sample, g. |
| Pf | = | Final absolute pressure of flask, mm Hg (in. Hg). |
| Pi | = | Initial absolute pressure of flask, mm Hg (in. Hg). |
| Pstd | = | Standard absolute pressure, 760 mm Hg (29.92 in. Hg). |
| RE | = | Relative error for QA audit samples, percent. |
| Tf | = | Final absolute temperature of flask, K (R). |
| Ti | = | Initial absolute temperature of flask, K (R). |
| Tstd | = | Standard absolute temperature, 293 K (528 R). |
| Vsc | = | Sample volume at standard conditions (dry basis), ml. |
| Vf | = | Volume of flask and valve, ml. |
| Va | = | Volume of absorbing solution, 25 ml. |
12.2 Spectrophotometer calibration Factor.
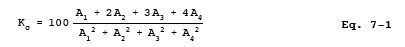
12.3 Sample Volume, Dry Basis, Corrected to Standard Conditions.
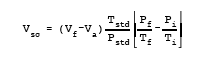
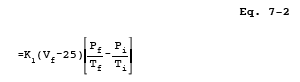
where:
| K1 | = | 0.3858 K/mm Hg for metric units, |
| = | 17.65 R/in. Hg for English units. |
12.4 Total g NO2 per sample.
m = 2 Kc A F Eq. 7-3
where:
| 2 | = | 50/25, the aliquot factor. |
NOTE: If other than a 25-ml aliquot is used for analysis, the factor 2 must be replaced by a corresponding factor.
12.5 Sample Concentration, Dry Basis, Corrected to Standard Conditions.
C = K2 (m/Vsc) Eq. 7-4
where:
| K2 | = | 103 (mg/m3)/(g/ml) for metric units, |
| = | 6.242 x 10-5 (lb/scf)/(g/ml) for English units. |
12.6 Relative Error for QA Audit Samples.
RE = 100 (Cd - Ca)/C>a Eq. 7-5
13.0 Method Performance.
13.1 Range.
The analytical range of the method has been determined to be 2 to 400 milligrams NOx (as NO2) per dry standard cubic meter, without having to dilute the sample.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
1. Standard Methods of Chemical Analysis. 6th ed. New York, D. Van Nostrand Co., Inc. 1962. Vol. 1, pp. 329-330.
2. Standard Method of Test for Oxides of Nitrogen in Gaseous Combustion Products (Phenoldisulfonic Acid Procedure). In: 1968 Book of ASTM Standards, Part 26. Philadelphia, PA. 1968. ASTM Designation D 1608-60, pp. 725-729.
3. Jacob, M.B. The Chemical Analysis of Air Pollutants. New York. Interscience Publishers, Inc. 1960. Vol. 10, pp. 351-356.
4. Beatty, R.L., L.B. Berger, and H.H. Schrenk. Determination of Oxides of Nitrogen by the Phenoldisulfonic Acid Method. Bureau of Mines, U.S. Dept. of Interior. R.I. 3687. February 1943.
5. Hamil, H.F. and D.E. Camann. Collaborative Study of Method for the Determination of Nitrogen Oxide Emissions from Stationary Sources (Fossil Fuel-Fired Steam Generators). Southwest Research Institute Report for Environmental Protection Agency. Research Triangle Park, NC. October 5, 1973.
6. Hamil, H.F. and R.E. Thomas. Collaborative Study of Method for the Determination of Nitrogen Oxide Emissions from Stationary Sources (Nitric Acid Plants). Southwest Research Institute Report for Environmental Protection Agency. Research Triangle Park, NC. May 8, 1974.
7. Stack Sampling Safety Manual (Draft). U.S. Environmental Protection Agency, Office of Air Quality Planning and Standards, Research Triangle Park, NC. September 1978.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
Figure 7-1. Sampling Train, Flask Valve, and Flask.
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources