- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalThe EPA Administrator, Gina McCarthy, signed the following notice on 9/29/2015, and EPA is submitting it for publication in the Federal Register (FR). While we have taken steps to ensure the accuracy of this Internet version of the rule, it is not the official version of the rule for purposes of compliance. Please refer to the official version in a forthcoming FR publication, which will appear on the Government Printing Office's FDSys website (https://gpo.gov/fdsys/search/home.action) and on Regulations.gov (https://www.regulations.gov) in Docket No. EPA- HQ-OAR-2010-0682. Once the official version of this document is published in the FR, this version will be removed from the Internet and replaced with a link to the official version
Method 325B—Volatile Organic Compounds from Fugitive and Area Sources:
Sampler Preparation and Analysis
1.1 Scope and Application
1.2 This method describes thermal desorption / gas chromatography (TD/GC) analysis of volatile organic compounds (VOCs) from fugitive and area emission sources collected onto sorbent tubes using passive sampling. It could also be applied to the TD/GC analysis of VOCs collected using active (pumped) sampling onto sorbent tubes. The concentration of airborne VOCs at or near potential fugitive- or area-emission sources may be determined using this method in combination with Method 325A. Companion Method 325A (Sampler Deployment and VOC Sample Collection) describes procedures for deploying the sorbent tubes and passively collecting VOCs.
1.3 The preferred GC detector for this method is a mass spectrometer (MS), but flame ionization detectors (FID) may also be used. Other conventional GC detectors such as electron
capture (ECD), photoionization (PID), or flame photometric (FPD) may also be used if they are selective and sensitive to the target compound(s) and if they meet the method performance criteria provided in this method.
1.4 There are 97 VOCs listed as hazardous air pollutants in Title III of the Clean Air Act Amendments of 1990. Many of these VOC are candidate compounds for this method. Compounds with known uptake rates for CarbographTM 1 TD, CarbopackTM B, or CarbopackTM X are listed in Table 12.1. This method provides performance criteria to demonstrate acceptable performance of the method (or modifications of the method) for monitoring one or more of the compounds listed Table 12.1. If standard passive sampling tubes are packed with other sorbents or used for other analytes than those listed in Table 12.1, then method performance and relevant uptake rates should be verified according to Addendum A to this method or by one of the following national/international standard methods: ISO 16017- 2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN 14662- 4:2005 (all incorporated by reference—see �63.14), or reported in the peer-reviewed open literature.
1.5 The analytical approach using TD/GC/MS is based on previously published EPA guidance in Compendium Method TO-17 (https://www.epa.gov/ttnamti1/airtox.html#compendium) (Reference
1), which describes active (pumped) sampling of VOCs from
ambient air onto tubes packed with thermally stable adsorbents.
1.6 Inorganic gases not suitable for analysis by this method include oxides of carbon, nitrogen and sulfur, ozone (O3), and other diatomic permanent gases. Other pollutants not suitable for this analysis method include particulate pollutants, (i.e., fumes, aerosols, and dusts), compounds too
labile (reactive) for conventional GC analysis, and VOCs that are more volatile than propane.
2.1 Summary of Method
2.2 This method provides procedures for the preparation, conditioning, blanking, and shipping of sorbent tubes prior to sample collection.
2.3 Laboratory and field personnel must have experience of sampling trace-level VOCs using sorbent tubes (References 2,5) and must have experience operating thermal desorption/GC/multi- detector instrumentation.
2.4 Key steps of this method as implemented for each sample tube include: Stringent leak testing under stop flow, recording ambient temperature conditions, adding internal standards, purging the tube, thermally desorbing the sampling tube, refocusing on a focusing trap, desorbing and transferring/injecting the VOCs from the secondary trap into the capillary GC column for separation and analysis.
2.5 Water management steps incorporated into this method
include: a) selection of hydrophobic sorbents in the sampling tube; b) optional dry purging of sample tubes prior to analysis; and c) additional selective elimination of water during primary (tube) desorption (if required) by selecting trapping sorbents and temperatures such that target compounds are quantitatively retained while water is purged to vent.
3.1 Definitions
(See also Section 3.0 of Method 325A).
3.2 Blanking is the desorption and confirmatory analysis
of conditioned sorbent tubes before they are sent for field sampling.
3.3 Breakthrough volume and associated relation to passive
sampling. Breakthrough volumes, as applied to active sorbent
tube sampling, equate to the volume of air containing a constant concentration of analyte that may be passed through a sorbent tube at a given temperature before a detectable level (5 percent) of the input analyte concentration elutes from the tube. Although breakthrough volumes are directly related to active rather than passive sampling, they provide a measure of the strength of the sorbent-sorbate interaction and therefore also relate to the efficiency of the passive sampling process.
The best direct measure of passive sampling efficiency is the stability of the uptake rate. Quantitative passive sampling is compromised when the sorbent no longer acts as a perfect sink –
i.e., when the concentration of a target analyte immediately
above the sorbent sampling surface no longer approximates to zero. This causes a reduction in the uptake rate over time. If the uptake rate for a given analyte on a given sorbent tube remains relatively constant — i.e., if the uptake rate
determined for 48 hours is similar to that determined for 7 or
14 days—the user can be confident that passive sampling is occurring at a constant rate. As a general rule of thumb, such ideal passive sampling conditions typically exist for analyte:sorbent combinations where the breakthrough volume exceeds 100 L (Reference 4).
3.4 Continuing calibration verification sample (CCV).
Single level calibration samples run periodically to confirm that the analytical system continues to generate sample results within acceptable agreement to the current calibration curve.
3.5 Focusing trap is a cooled, secondary sorbent trap
integrated into the analytical thermal desorber. It typically has a smaller i.d. and lower thermal mass than the original sample tube allowing it to effectively refocus desorbed analytes and then heat rapidly to ensure efficient transfer/injection into the capillary GC analytical column.
3.6 High Resolution Capillary Column Chromatography uses
fused silica capillary columns with an inner diameter of 320 μm or less and with a stationary phase film thickness of 5 μm or
less.
3.7 h is time in hours.
3.8 i.d. is inner diameter.
3.9 min is time in minutes.
3.10 Method Detection Limit is the lowest level of analyte
that can be detected in the sample matrix with 99% confidence.
3.11 MS-SCAN is the mode of operation of a GC quadrupole
mass spectrometer detector that measures all ions over a given mass range over a given period of time.
3.12 MS-SIM is the mode of operation of a GC quadrupole
mass spectrometer detector that measures only a single ion or a selected number of discrete ions for each analyte.
3.13 o.d. is outer diameter.
3.14 ppbv is parts per billion by volume.
3.15 Thermal desorption is the use of heat and a flow of
inert (carrier) gas to extract volatiles from a solid matrix. No solvent is required.
3.16 Total ion chromatogram is the chromatogram produced
from a mass spectrometer detector collecting full spectral information.
3.17 Two-stage thermal desorption is the process of
thermally desorbing analytes from a sorbent tube, reconcentrating them on a focusing trap (see Section 3.4), which is then itself rapidly heated to �inject� the concentrated
compounds into the GC analyzer.
3.18 VOC is volatile organic compound.
4.1 Analytical Interferences
4.2 Interference from Sorbent Artifacts. Artifacts may
include target analytes as well as other VOC that co-elute chromatographically with the compounds of interest or otherwise interfere with the identification or quantitation of target analytes.
4.2.1 Sorbent decomposition artifacts are VOCs that form when sorbents degenerate, e.g., when exposed to reactive species
during sampling. For example, benzaldehyde, phenol, and acetophenone artifacts are reported to be formed via oxidation of the polymeric sorbent Tenax� when sampling high concentration (100-500 ppb) ozone atmospheres (Reference 5).
4.2.2 Preparation and storage artifacts are VOCs that were not completely cleaned from the sorbent tube during conditioning or that are an inherent feature of that sorbent at a given temperature.
4.2 Humidity. Moisture captured during sampling can
interfere with VOC analysis. Passive sampling using tubes packed with hydrophobic sorbents, like those described in this method, minimizes water retention. However, if water interference is found to be an issue under extreme conditions, one or more of the water management steps described in Section 2.4 can be
applied.
4.3 Contamination from Sample Handling. The type of
analytical thermal desorption equipment selected should exclude the possibility of outer tube surface contamination entering the sample flow path (see Section 6.6). If the available system does not meet this requirement, sampling tubes and caps must be handled only while wearing clean, white cotton or powder free nitrile gloves to prevent contamination with body oils, hand lotions, perfumes, etc.
5.1 Safety
5.2 This method does not address all of the safety concerns associated with its use. It is the responsibility of the user of this standard to establish appropriate field and laboratory safety and health practices prior to use.
5.3 Laboratory analysts must exercise extreme care in working with high-pressure gas cylinders.
5.4 Due to the high temperatures involved, operators must use caution when conditioning and analyzing tubes.
6.1 Equipment and Supplies
6.2 Tube Dimensions and Materials. The sampling tubes for
this method are 3.5-inches (89 mm) long, 1/4 inch (6.4 mm) o.d., and 5 mm i.d. passive sampling tubes (see Figure 6.1). The tubes are made of inert-coated stainless steel with the central section (up to 60 mm) packed with sorbent, typically supported
between two 100 mesh stainless steel gauze. The tubes have a cross sectional area of 19.6 square mm (5 mm i.d.). When used for passive sampling, these tubes have an internal diffusion (air) gap (DG) of 1.5 cm between the sorbent retaining gauze at the sampling end of the tube, and the gauze in the diffusion cap.
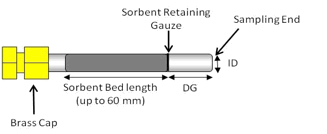
Figure 6.1. Cross Section View of Passive Sorbent Tube
6.3 Tube Conditioning Apparatus.
6.3.1 Freshly packed or newly purchased tubes must be conditioned as described in Section 9 using an appropriate dedicated tube conditioning unit or the thermal desorber. Note that the analytical TD system should be used for tube conditioning if it supports a dedicated tube conditioning mode in which effluent from contaminated tubes is directed to vent without passing through key parts of the sample flow path such as the focusing trap.
6.3.2 Dedicated tube conditioning units must be leak-tight to prevent air ingress, allow precise and reproducible temperature selection (�5C), offer a temperature range at least
as great as that of the thermal desorber, and support inert gas flows in the range up to 100 mL/min.
Note: For safety and to avoid laboratory contamination, effluent gases from freshly packed or highly contaminated tubes should be passed through a charcoal filter during the conditioning process to prevent desorbed VOCs from polluting the laboratory atmosphere.
6.4 Tube Labeling.
6.4.1 Label the sample tubes with a unique permanent identification number and an indication of the sampling end of the tube. Labeling options include etching and TD-compatible electronic (radio frequency identification (RFID)) tube labels.
6.4.2 To avoid contamination, do not make ink markings of any kind on clean sorbent tubes or apply adhesive labels.
Note: TD-compatible electronic (RFID) tube labels are available commercially and are compatible with some brands of thermal desorber. If used, these may be programmed with relevant tube and sample information, which can be read and automatically transcribed into the sequence report by the TD system (see Section 8.6 of Method 325A).
6.4 Blank and Sampled Tube Storage Apparatus
6.4.1 Long-term storage caps. Seal clean, blank and sampled sorbent tubes using inert, long-term tube storage caps comprising non-greased, 2-piece, 0.25-inch, metal SwageLok�-type
screw caps fitted with combined polytetrafluoroethylene ferrules.
6.4.2 Storage and transportation containers. Use clean glass jars, metal cans or rigid, non-emitting polymer boxes.
Note: You may add a small packet of new activated charcoal or charcoal/silica gel to the shipping container for storage and transportation of batches of conditioned sorbent tubes prior to use. Coolers without ice packs make suitable shipping boxes for containers of tubes because the coolers help to insulate the samples from extreme temperatures (e.g., if left in a parked
vehicle).
6.5 Unheated GC Injection Unit for Loading Standards onto
Blank Tubes. A suitable device has a simple push fit or finger-
tightening connector for attaching the sampling end of blank sorbent tubes without damaging the tube. It also has a means of controlling carrier gas flow through the injector and attached sorbent tube at 50-100 mL/min and includes a low emission septum cap that allows the introduction of gas or liquid standards via appropriate syringes. Reproducible and quantitative transfer of higher boiling compounds in liquid standards is facilitated if the injection unit allows the tip of the syringe to just touch the sorbent retaining gauze inside the tube.
6.6 Thermal Desorption Apparatus. The manual or automated
thermal desorption system must heat sorbent tubes while a
controlled flow of inert (carrier) gas passes through the tube and out of the sampling end. The apparatus must also incorporate a focusing trap to quantitatively refocus compounds desorbed from the tube. Secondary desorption of the focusing trap should be fast/efficient enough to transfer the compounds into the high resolution capillary GC column without band broadening and without any need for further pre- or on-column focusing. Typical TD focusing traps comprise small sorbent traps (Reference 16) that are electrically-cooled using multistage Peltier cells (References 17, 18). The direction of gas flow during trap desorption should be the reverse of that used for focusing to extend the compatible analyte volatility range. Closed cycle coolers offer another cryogen-free trap cooling option. Other TD system requirements and operational stages are described in Section 11 and in Figures 17-2 through 17-4.
6.7 Thermal Desorber - GC Interface.
6.7.1 The interface between the thermal desorber and the GC must be heated uniformly and the connection between the transfer line insert and the capillary GC analytical column itself must be leak tight.
6.7.2 A portion of capillary column can alternatively be threaded through the heated transfer line / TD interface and connected directly to the thermal desorber.
Note: Use of a metal syringe-type needle or unheated length
of fused silica pushed through the septum of a conventional GC injector is not permitted as a means of interfacing the thermal desorber to the chromatograph. Such connections result in cold spots, cause band broadening and are prone to leaks.
6.8 GC/MS Analytical Components.
6.8.1 The GC system must be capable of temperature programming and operation of a high resolution capillary column. Depending on the choice of column (e.g., film thickness) and the
volatility of the target compounds, it may be necessary to cool the GC oven to subambient temperatures (e.g., -50 C) at the
start of the run to allow resolution of very volatile organic compounds.
6.8.2 All carrier gas lines supplying the GC must be constructed from clean stainless steel or copper tubing. Non- polytetrafluoroethylene thread sealants. Flow controllers, cylinder regulators, or other pneumatic components fitted with rubber components are not suitable.
6.9 Chromatographic Columns. High-resolution, fused silica
or equivalent capillary columns that provide adequate separation of sample components to permit identification and quantitation of target compounds must be used.
Note: 100-percent methyl silicone or 5-percent phenyl, 95- percent methyl silicone fused silica capillary columns of 0.25- to 0.32-mm i.d. of varying lengths and with varying thicknesses
of stationary phase have been used successfully for non-polar and moderately polar compounds. However, given the diversity of potential target lists, GC column choice is left to the operator, subject to the performance criteria of this method.
6.10 Mass Spectrometer. Linear quadrupole, magnetic
sector, ion trap or time-of-flight mass spectrometers may be used provided they meet specified performance criteria. The mass detector must be capable of collecting data from 35 to 300 atomic mass units (amu) every 1 second or less, utilizing 70 volts (nominal) electron energy in the electron ionization mode, and producing a mass spectrum that meets all the instrument performance acceptance criteria in Section 9 when 50 g or less of p-bromofluorobenzene is analyzed.
7.1 Reagents and Standards
7.2 Sorbent Selection.
7.2.1 Use commercially packed tubes meeting the requirements of this method or prepare tubes in the laboratory using sieved sorbents of particle size in the range 20 to 80 mesh that meet the retention and quality control requirements of this method.
7.2.2 This passive air monitoring method can be used without the evaluation specified in Addendum A if the type of tubes described in Section 6.1 are packed with 4-6 cm (typically 400-650 mg) of the sorbents listed in Table 12.1 and used for
the respective target analytes.
Note: Although CarbopackTM X is the optimum sorbent choice for passive sampling of 1,3-butadiene, recovery of compounds with vapor pressure lower than benzene may be difficult to achieve without exceeding sorbent maximum temperature limitations (see Table 8.1). See ISO 16017-2:2003(E) or ASTM D6196-03 (Reapproved 2009) (both incorporated by reference—see
�63.14) for more details on sorbent choice for air monitoring using passive sampling tubes.
7.2.3 If standard passive sampling tubes are packed with other sorbents or used for analytes other than those tabulated in Section 12.0, method performance and relevant uptake rates should be verified according to Addendum A to this method or by following the techniques described in one of the following national/international standard methods: ISO 16017-2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN 14662-4:2005 (all incorporated by reference—see �63.14) – or reported in the peer- reviewed open literature. A summary table and the supporting evaluation data demonstrating the selected sorbent meets the requirements in Addendum A to this method must be submitted to the regulatory authority as part of a request to use an alternative sorbent.
7.2.4 Passive (diffusive) sampling and thermal desorption methods that have been evaluated at relatively high atmospheric
concentrations (i.e., mid-ppb to ppm) and published for use in
workplace air and industrial/mobile source emissions testing (References 9-20) may be applied to this procedure. However, the validity of any shorter term uptake rates must be verified and adjusted if necessary for the longer monitoring periods required by this method by following procedures described in Addendum A to this method or those presented in national/international standard methods: ISO 16017-2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN 14662-4:2005 (all incorporated by reference-see
�63.14).
7.2.5 Suitable sorbents for passive sampling must have breakthrough volumes of at least 20 L (preferably >100 L) for the compounds of interest and must quantitatively release the analytes during desorption without exceeding maximum temperatures for the sorbent or instrumentation.
7.2.6 Repack/replace the sorbent tubes or demonstrate tube performance following the requirements in Addendum A to this method at least every 2 years or every 50 uses, whichever occurs first.
7.2 Gas Phase Standards.
7.2.1 Static or dynamic standard atmospheres may be used to prepare calibration tubes and/or to validate passive sampling uptake rates and can be generated from pure chemicals or by diluting concentrated gas standards. The standard atmosphere
must be stable at ambient pressure and accurate to �10 percent of the target gas concentration. It must be possible to maintain standard atmosphere concentrations at the same or lower levels than the target compound concentration objectives of the test.
Test atmospheres used for validation of uptake rates must also contain at least 35 percent relative humidity.
Note: Accurate, low-(ppb-) level gas-phase VOC standards are difficult to generate from pure materials and may be unstable depending on analyte polarity and volatility. Parallel monitoring of vapor concentrations with alternative methods, such as pumped sorbent tubes or sensitive/selective on-line detectors, may be necessary to minimize uncertainty. For these reasons, standard atmospheres are rarely used for routine calibration.
7.2.2 Concentrated, pressurized gas phase standards.
Accurate (�5 percent or better), concentrated gas phase standards supplied in pressurized cylinders may also be used for calibration. The concentration of the standard should be such that a 0.5 - 5.0 mL volume contains approximately the same mass of analytes as will be collected from a typical air sample.
7.2.3 Follow manufacturer�s guidelines concerning storage conditions and recertification of the concentrated gas phase standard. Gas standards must be recertified a minimum of once every 12 months.
7.3 Liquid Standards. Target analytes can also be
introduced to the sampling end of sorbent tubes in the form of liquid calibration standards.
7.3.1 The concentration of liquid standards must be such that an injection of 0.5-2 �l of the solution introduces the same mass of target analyte that is expected to be collected during the passive air sampling period.
7.3.2 Solvent Selection. The solvent selected for the
liquid standard must be pure (contaminants <10 percent of minimum analyte levels) and must not interfere chromatographically with the compounds of interest.
7.3.3 If liquid standards are sourced commercially, follow manufacturer�s guidelines concerning storage conditions and shelf life of unopened and opened liquid stock standards.
Note: Commercial VOC standards are typically supplied in volatile or non-interfering solvents such as methanol.
7.3.4 Working standards must be stored at 6 C or less and used or discarded within two weeks of preparation.
7.4 Gas Phase Internal Standards.
7.4.1 Gas-phase deuterated or fluorinated organic compounds may be used as internal standards for MS-based systems.
7.4.2 Typical compounds include deuterated toluene, perfluorobenzene and perfluorotoluene.
7.4.3 Use multiple internal standards to cover the volatility range of the target analytes.
7.4.4 Gas-phase standards must be obtained in pressurized cylinders and containing vendor certified gas concentrations accurate to �5 percent. The concentration should be such that the mass of internal standard components introduced is similar to those of the target analytes collected during field monitoring.
7.5 Preloaded Standard Tubes. Certified, preloaded
standard tubes, accurate within �5 percent for each analyte at the microgram level and �10 percent at the nanogram level, are available commercially and may be used for auditing and quality control purposes. (See Section 9.5 for audit accuracy evaluation criteria.) Certified preloaded tubes may also be used for routine calibration.
Note: Proficiency testing schemes are also available for TD/GC/MS analysis of sorbent tubes preloaded with common analytes such as benzene, toluene, and xylene.
7.6 Carrier Gases. Use inert, 99.999-percent or higher
purity helium as carrier gas. Oxygen and organic filters must be installed in the carrier gas lines supplying the analytical system according to the manufacturer�s instructions. Keep records of filter and oxygen scrubber replacement.
8.1 Sorbent Tube Handling (Before and After Sampling)
8.2 Sample Tube Conditioning.
8.2.1 Sampling tubes must be conditioned using the apparatus described in Section 6.2.
8.2.2 New tubes should be conditioned for 2 hours to supplement the vendor�s conditioning procedure. Recommended temperatures for tube conditioning are given in Table 8.1.
8.2.3 After conditioning, the blank must be verified on each new sorbent tube and on 10 percent of each batch of reconditioned tubes. See Section 9.0 for acceptance criteria.
Table 8.1 Example Sorbent Tube Conditioning Parameters
Sampling Sorbent |
Maximum Temperature (C) |
Conditioning Temperature (C) |
Carrier Gas Flow Rate |
Carbotrap C CarbopackTM C Anasorb GCB2 CarbographTM 1 TD Carbotrap CarbopackTM B Anasorb GCB1 |
>400 |
350 |
100 mL/min |
Tenax TA CarbopackTM X |
350 |
330 |
100 mL/min |
8.2 Capping, Storage and Shipment of Conditioned Tubes.
8.2.1 Conditioned tubes must be sealed using long-term storage caps (see Section 6.4) pushed fully down onto both ends of the PS sorbent tube, tightened by hand and then tighten an additional quarter turn using an appropriate tool.
8.2.2 The capped tubes must be kept in appropriate containers for storage and transportation (see Section 6.4.2).
Containers of sorbent tubes may be stored and shipped at ambient temperature and must be kept in a clean environment.
8.2.3 You must keep batches of capped tubes in their shipping boxes or wrap them in uncoated aluminum foil before placing them in their storage container, especially before air freight, because the packaging helps hold caps in position if the tubes get very cold.
8.3 Calculating the Number of Tubes Required for a
Monitoring Exercise.
8.3.1 Follow guidance given in Method 325A to determine the number of tubes required for site monitoring.
8.3.2 The following additional samples will also be required: Laboratory blanks as specified in Section 9.1.2 (one per analytical sequence minimum), field blanks as specified in Section 9.3.2 (two per sampling period minimum), CCV tubes as specified in Section 10.9.4. (at least one per analysis sequence or every 24 hours), and duplicate samples as specified in Section 9.4 (at least one duplicate sample is required for every
10 sampling locations during each monitoring period).
8.4 Sample Collection.
8.4.1 Allow the tubes to equilibrate with ambient temperature (approximately 30 minutes to 1 hour) at the monitoring location before removing them from their storage/shipping container for sample collection.
8.4.2 Tubes must be used for sampling within 30 days of conditioning (Reference 4).
8.4.3 During field monitoring, the long-term storage cap at the sampling end of the tube is replaced with a diffusion cap and the whole assembly is arranged vertically, with the sampling end pointing downward, under a protective hood or shield – See Section 6.1 of Method 325A for more details.
8.5 Sample Storage.
8.5.1 After sampling, tubes must be immediately resealed with long-term storage caps and placed back inside the type of storage container described in Section 6.4.2.
8.5.2 Exposed tubes may not be placed in the same container as clean tubes. They should not be taken back out of the container until ready for analysis and after they have had time to equilibrate with ambient temperature in the laboratory.
8.5.3 Sampled tubes must be inspected before analysis to identify problems such as loose or missing caps, damaged tubes, tubes that appear to be leaking sorbent or container contamination. Any and all such problems must be documented together with the unique identification number of the tube or tubes concerned. Affected tubes must not be analyzed but must be set aside.
8.5.4 Intact tubes must be analyzed within 30 days of the end of sample collection (within one week for limonene, carene,
bis-chloromethyl ether, labile sulfur or nitrogen-containing compounds, and other reactive VOCs).
Note: Ensure ambient temperatures stay below 23 �C during transportation and storage. Refrigeration is not normally required unless the samples contain reactive compounds or cannot be analyzed within 30 days. If refrigeration is used, the atmosphere inside the refrigerator must be clean and free of organic solvents.
9.1 Quality Control
9.2 Laboratory Blank. The analytical system must be
demonstrated to be contaminant free by performing a blank analysis at the beginning of each analytical sequence to demonstrate that the secondary trap and TD/GC/MS analytical equipment are free of any significant interferents.
9.2.1 Laboratory blank tubes must be prepared from tubes that are identical to those used for field sampling.
9.2.2 Analysis of at least one laboratory blank is required per analytical sequence. The laboratory blank must be stored in the laboratory under clean, controlled ambient temperature conditions.
9.2.3 Laboratory blank/artifact levels must meet the requirements of Section 9.2.2 (see also Table 17.1). If the laboratory blank does not meet requirements, stop and perform corrective actions and then re-analyze laboratory blank to
ensure it meets requirements.
9.2 Tube Conditioning.
9.2.1 Conditioned tubes must be demonstrated to be free of contaminants and interference by running 10 percent of the blank tubes selected at random from each conditioned batch under standard sample analysis conditions (see Section 8.1).
9.2.2 Confirm that artifacts and background contamination are � 0.2 ppbv or less than three times the detection limit of the procedure or less than 10 percent of the target compound(s) mass that would be collected if airborne concentrations were at the regulated limit value, whichever is larger. Only tubes that meet these criteria can be used for field monitoring, field or laboratory blanks, or for system calibration.
9.2.3 If unacceptable levels of VOCs are observed in the tube blanks, then the processes of tube conditioning and checking the blanks must be repeated.
9.3 Field Blanks.
9.3.1 Field blank tubes must be prepared from tubes that are identical to those used for field sampling – i.e., they
should be from the same batch, have a similar history, and be conditioned at the same time.
9.3.2 Field blanks must be shipped to the monitoring site with the sampling tubes and must be stored at the sampling location throughout the monitoring exercise. The field blanks
must be installed under a protective hood/cover at the sampling location, but the long-term storage caps must remain in place throughout the monitoring period (see Method 325A). The field blanks are then shipped back to the laboratory in the same container as the sampled tubes. One field blank tube is required for every 10 sampled tubes on a monitoring exercise and no less than two field blanks should be collected, regardless of the size of the monitoring study.
9.3.3 Field blanks must contain no greater than one-third of the measured target analyte or compliance limit for field samples (see Table 17.1). If either field blank fails, flag all data that do not meet this criterion with a note that the associated results are estimated and likely to be biased high due to field blank background.
9.4 Duplicate Samples. Duplicate (co-located) samples
collected must be analyzed and reported as part of method quality control. They are used to evaluate sampling and analysis precision. Relevant performance criteria are given in Section 9.9.
9.5 Method Performance Criteria. Unless otherwise noted,
monitoring method performance specifications must be demonstrated for the target compounds using the procedures described in Addendum A to this method and the statistical approach presented in Method 301.
9.6 Method Detection Limit. Determine the method detection
limit under the analytical conditions selected (see Section 11.3) using the procedure in Section 15 of Method 301. The method detection limit is defined for each system by making seven replicate measurements of a concentration of the compound of interest within a factor of five of the detection limit.
Compute the standard deviation for the seven replicate concentrations, and multiply this value by three. The results should demonstrate that the method is able to detect analytes such as benzene at concentrations as low as 50 ppt or 1/3rd (preferably 1/10th) of the lowest concentration of interest, whichever is larger.
Note: Determining the detection limit may be an iterative process as described in 40 CFR part 136, Appendix B.
9.7 Analytical Bias. Analytical bias must be demonstrated
to be within �30 percent using Equation 9.1. Analytical bias must be demonstrated during initial setup of this method and as part of the CCV carried out with every sequence of 10 samples or less (see Section 9.14). Calibration standard tubes (see Section 10.0) may be used for this purpose.
AnalyticalBias
SpikedValue MeasuredValue SpikedValue
![]() 100
100
Eq. 9.1
Where:
Spiked Value = A known mass of VOCs added to the tube.
Measured Value = Mass determined from analysis of the tube.
9.8 Analytical Precision. Demonstrate an analytical
precision within �20 percent using Equation 9.2. Analytical precision must be demonstrated during initial setup of this method and at least once per year. Calibration standard tubes may be used (see Section 10.0) and data from CCV may also be applied for this purpose.
Analytical Precision
100
![]() A
A
Eq. 9.2
Where:
A1 = A measurement value taken from one spiked tube.
A2 = A measurement value taken from a second spiked tube.
A = The average of A1 and A2.
9.9 Field Replicate Precision. Use Equation 9.3 to
determine and report replicate precision for duplicate field samples (see Section 9.4). The level of agreement between duplicate field samples is a measure of the precision achievable for the entire sampling and analysis procedure. Flag data sets for which the duplicate samples do not agree within 30 percent.
Field Precision
100
![]() F
F
Eq. 9.3
Where:
F1 = A measurement value (mass) taken from one of the two field replicate tubes used in sampling.
F2 = A measurement value (mass) taken from the second of two field replicate tubes used in sampling.
F = The average of F1 and F2.
9.10 Desorption Efficiency and Compound Recovery. The
efficiency of the thermal desorption method must be determined.
9.10.1 Quantitative (>95 percent) compound recovery must be demonstrated by repeat analyses on a same standard tube.
9.10.2 Compound recovery through the TD system can also be demonstrated by comparing the calibration check sample response factor obtained from direct GC injection of liquid standards with that obtained from thermal desorption analysis response factor using the same column under identical conditions.
9.10.3 If the relative response factors obtained for one or more target compounds introduced to the column via thermal desorption fail to meet the criteria in Section 9.10.1, you must adjust the TD parameters to meet the criteria and repeat the experiment. Once the thermal desorption conditions have been optimized, you must repeat this test each time the analytical system is recalibrated to demonstrate continued method performance.
9.11 Audit Samples. Certified reference standard samples
must be used to audit this procedure (if available). Accuracy within 30 percent must be demonstrated for relevant ambient air concentrations (0.5 to 25 ppb).
9.12 Mass Spectrometer Tuning Criteria. Tune the mass
spectrometer (if used) according to manufacturer�s specifications. Verify the instrument performance by analyzing a
50 g injection of bromofluorobenzene. Prior to the beginning of each analytical sequence or every 24 hours during continuous GC/MS operation for this method demonstrate that the bromofluorobenzene tuning performance criteria in Table 9.1 have been met.
Table 9.1 GC/MS Tuning Criteria1
Target Mass |
Rel. To Mass |
Lower Limit % |
Upper Limit % |
50 |
95 |
8 |
40 |
75 |
95 |
30 |
66 |
95 |
95 |
100 |
100 |
96 |
95 |
5 |
9 |
173 |
174 |
0 |
2 |
174 |
95 |
50 |
120 |
175 |
174 |
4 |
9 |
176 |
174 |
93 |
101 |
177 |
176 |
5 |
9 |
1 All ion abundances must be normalized to m/z 95, the nominal base peak, even though the ion abundance of m/z 174 may be up to 120 percent that of m/z 95.
9.13 Routine CCV at the Start of a Sequence. Run CCV
before each sequence of analyses and after every tenth sample to ensure that the previous multi-level calibration (see Section 10.6.3) is still valid.
9.13.1 The sample concentration used for the CCV should be near the mid-point of the multi-level calibration range.
9.13.2 Quantitation software must be updated with response
factors determined from the CCV standard. The percent deviation between the initial calibration and the CCV for all compounds must be within 30 percent.
9.14 CCV at the End of a Sequence. Run another CCV after
running each sequence of samples. The initial CCV for a subsequent set of samples may be used as the final CCV for a previous analytical sequence, provided the same analytical method is used and the subsequent set of samples is analyzed immediately (within 4 hours) after the last CCV.
9.15 Additional Verification. Use a calibration check
standard from a second, separate source to verify the original calibration at least once every three months.
9.16 Integration Method. Document the procedure used for
integration of analytical data including field samples, calibration standards and blanks.
9.17 QC Records. Maintain all QC reports/records for each
TD/GC/MS analytical system used for application of this method. Routine quality control requirements for this method are listed below and summarized in Table 17.1.
10.1 Calibration and Standardization
10.2 Calibrate the analytical system using standards covering the range of analyte masses expected from field samples.
10.3 Analytical results for field samples must fall within
the calibrated range of the analytical system to be valid.
10.4 Calibration standard preparation must be fully traceable to primary standards of mass and/or volume, and/or be confirmed using an independent certified reference method.
10.4.1 Preparation of calibration standard tubes from standard atmospheres.
10.4.1.1 Subject to the requirements in Section 7.2.1, low-level standard atmospheres may be introduced to clean, conditioned sorbent tubes in order to produce calibration standards.
10.4.1.2 The standard atmosphere generator or system must be capable of producing sufficient flow at a constant rate to allow the required analyte mass to be introduced within a reasonable time frame and without affecting the concentration of the standard atmosphere itself.
10.4.1.3 The sampling manifold may be heated to minimize risk of condensation but the temperature of the gas delivered to the sorbent tubes may not exceed 100 F.
10.4.1.4 The flow rates passed through the tube should be in the order of 50-100 mL/min and the volume of standard atmosphere sampled from the manifold or chamber must not exceed the breakthrough volume of the sorbent at the given temperature.
10.4 Preparation of calibration standard tubes from concentrated gas standards.
10.4.1 If a suitable concentrated gas standard (see Section 7.2.2) can be obtained, follow the manufacturer�s recommendations relating to suitable storage conditions and product lifetime.
10.4.2 Introduce precise 0.5 to 500.0 mL aliquots of the standard to the sampling end of conditioned sorbent tubes in a 50-100 mL/min flow of pure carrier gas.
Note: This can be achieved by connecting the sampling end of the tube to an unheated GC injector (see Section 6.6) and introducing the aliquot of gas using a suitable gas syringe. Gas sample valves could alternatively be used to meter the standard gas volume.
10.4.3 Each sorbent tube should be left connected to the flow of gas for 2 minutes after standard introduction. As soon as each spiked tube is removed from the injection unit, seal it with long-term storage caps and place it in an appropriate tube storage/transportation container if it is not to be analyzed within 24 hours.
10.5 Preparation of calibration standard tubes from liquid standards.
10.5.1 Suitable standards are described in Section 7.3.
10.5.2 Introduce precise 0.5 to 2 �l aliquots of liquid standards to the sampling end of sorbent tubes in a flow (50-100 mL/min) of carrier gas using a precision syringe and an unheated
injector (Section 6.5). The flow of gas should be sufficient to completely vaporize the liquid standard.
Note: If the analytes of interest are higher boiling than n-decane, reproducible analyte transfer to the sorbent bed is optimized by allowing the tip of the syringe to gently touch the sorbent retaining gauze at the sampling end of the tube.
10.5.3 Each sorbent tube is left connected to the flow of gas for 5 minutes after liquid standard introduction.
10.5.3.1 As soon as each spiked tube is removed from the injection unit, seal it with long-term storage caps and place it in an appropriate tube storage container if it is not to be analyzed within 24 hours.
Note: In cases where it is possible to selectively purge the solvent from the tube while all target analytes are quantitatively retained, a larger 2 �L injection may be made for optimum accuracy. However, if the solvent cannot be selectively purged and will be present during analysis, the injection volume should be as small as possible (e.g., 0.5 �L) to minimize
solvent interference.
Note: This standard preparation technique requires the entire liquid plug including the tip volume be brought into the syringe barrel. The volume in the barrel is recorded, the syringe is inserted into the septum of the spiking apparatus.
The liquid is then quickly injected. Any remaining liquid in the
syringe tip is brought back into the syringe barrel. The volume in the barrel is recorded and the amount spiked onto the tube is the difference between the before spiking volume and the after spiking volume. A bias occurs with this method when sample is drawn continuously up into the syringe to the specified volume and the calibration solution in the syringe tip is ignored.
10.6 Preparation of calibration standard tubes from multiple standards.
10.6.1 If it is not possible to prepare one standard containing all the compounds of interest (e.g., because of
chemical reactivity or the breadth of the volatility range), standard tubes can be prepared from multiple gas or liquid standards.
10.6.2 Follow the procedures described in Sections 10.4 and 10.5, respectively, for introducing each gas and/or liquid standard to the tube and load those containing the highest boiling compounds of interest first and the lightest species last.
10.7 Additional requirements for preparation of calibration tubes.
10.7.1 Storage of Calibration Standard Tubes
10.7.1.1 Seal tubes with long-term storage caps immediately after they have been disconnected from the standard loading manifold or injection apparatus.
10.7.1.2 Calibration standard tubes may be stored for no longer than 30 days and should be refrigerated if there is any risk of chemical interaction or degradation. Audit standards (see section 9.11) are exempt from this criteria and may be stored for the shelf-life specified on their certificates.
10.8 Keep records for calibration standard tubes to include the following:
10.8.1 The stock number of any commercial liquid or gas standards used.
10.8.2 A chromatogram of the most recent blank for each tube used as a calibration standard together with the associated analytical conditions and date of cleaning.
10.8.3 Date of standard loading.
10.8.4 List of standard components, approximate masses and associated confidence levels.
10.8.5 Example analysis of an identical standard with associated analytical conditions.
10.8.6 A brief description of the method used for standard preparation.
10.8.7 The standard�s expiration date.
10.9 TD/GC/MS using standard tubes to calibrate system response.
10.9.1 Verify that the TD/GC/MS analytical system meets the instrument performance criteria given in Section 9.1.
10.9.2 The prepared calibration standard tubes must be analyzed using the analytical conditions applied to field samples (see Section 11.0) and must be selected to ensure quantitative transfer and adequate chromatographic resolution of target compounds, surrogates, and internal standards in order to enable reliable identification and quantitation of compounds of interest. The analytical conditions should also be sufficiently stringent to prevent buildup of higher boiling, non-target contaminants that may be collected on the tubes during field monitoring.
10.9.3 Calibration range. Each TD/GC/MS system must be calibrated at five concentrations that span the monitoring range of interest before being used for sample analysis. This initial multi-level calibration determines instrument sensitivity under the analytical conditions selected and the linearity of GC/MS response for the target compounds. One of the calibration points must be within a factor of five of the detection limit for the compounds of interest.
10.9.4 One of the calibration points from the initial calibration curve must be at the same concentration as the daily CCV standard (e.g., the mass collected when sampling air at
typical concentrations).
10.9.5 Calibration frequency. Each GC/MS system must be recalibrated with a full 5-point calibration curve following
corrective action (e.g., ion source cleaning or repair, column
replacement) or if the instrument fails the daily calibration acceptance criteria.
10.9.5.1 CCV checks must be carried out on a regular routine basis as described in Section 9.14.
10.9.5.2 Quantitation ions for the target compounds are shown in Table 10.1. Use the primary ion unless interferences are present, in which case you should use a secondary ion.
Table 10.1 Clean Air Act Volatile Organic Compounds for Passive Sorbent Sampling
Compound |
CAS No. |
BP (C) |
Vapor pressure (mmHg)a |
MWb |
Characteristic Ion(s) |
|
Primary |
Secondary |
|||||
1,1-Dichloroethene |
75-35-4 |
32 |
500 |
96.9 |
61 |
96 |
3-Chloropropene |
107-05-1 |
44.5 |
340 |
76.5 |
76 |
41,39,78 |
1,1,2-Trichloro-1,2,2- trifluoroethane |
||||||
1,1-Dichloroethane |
75-34-3 |
57.0 |
230 |
99 |
63 |
65, 83, 85, 98, 100. |
1,2-Dichloroethane |
107-06-2 |
83.5 |
61.5 |
99 |
62 |
98 |
1,1,1-Trichloroethane |
71-55-6 |
74.1 |
100 |
133.4 |
97 |
99,61 |
Benzene |
71-43-2 |
80.1 |
76.0 |
78 |
78 |
|
Carbon tetrachloride |
56-23-5 |
76.7 |
90.0 |
153.8 |
117 |
119 |
1,2-Dichloropropane |
78-87-5 |
97.0 |
42.0 |
113 |
63 |
112 |
Trichloroethene |
79-01-6 |
87.0 |
20.0 |
131.4 |
95 |
97, 130, 132 |
1,1,2-Trichloroethane |
79-00-5 |
114 |
19.0 |
133.4 |
83 |
97, 85 |
Toluene |
108-88-3 |
111 |
22.0 |
92 |
92 |
91 |
Tetrachloroethene |
127-18-4 |
121 |
14.0 |
165.8 |
164 |
129, 131, 166 |
Chlorobenzene |
108-90-7 |
132 |
8.8 |
112.6 |
112 |
77, 114 |
Ethylbenzene |
100-41-4 |
136 |
7.0 |
106 |
91 |
106 |
m,p-Xylene |
108-38-3, 106-42-3 |
138 |
6.5 |
106.2 |
106 |
91 |
Compound |
CAS No. |
BP (C) |
Vapor pressure (mmHg)a |
MWb |
Characteristic Ion(s) |
|
Primary |
Secondary |
|||||
Styrene |
100-42-5 |
145 |
6.6 |
104 |
104 |
78 |
o-Xylene |
95-47-6 |
144 |
5.0 |
106.2 |
106 |
91 |
p-Dichlorobenzene |
106-46-7 |
173 |
0.60 |
147 |
146 |
111, 148 |
a Pressure in millimeters of mercury.
b Molecular weight.
11.1 Analytical Procedure
11.2 Preparation for Sample Analysis.
11.2.1 Each sequence of analyses must be ordered as follows:
11.2.1.1 CCV.
11.2.1.2 A laboratory blank.
11.2.1.3 Field blank.
11.2.1.4 Sample(s).
11.2.1.5 Field blank.
11.2.1.6 CCV after 10 field samples.
11.2.1.7 CCV at the end of the sample batch.
11.2 Pre-desorption System Checks and Procedures.
11.2.1 Ensure all sample tubes and field blanks are at ambient temperature before removing them from the storage container.
11.2.2 If using an automated TD/GC/MS analyzer, remove the long-term storage caps from the tubes, replace them with appropriate analytical caps, and load them into the system in
the sequence described in Section 11.1. Alternatively, if using a manual system, uncap and analyze each tube, one at a time, in the sequence described in Section 11.1.
11.2.3 The following thermal desorption system integrity checks and procedures are required before each tube is analyzed.
Note: Commercial thermal desorbers should implement these steps automatically.
11.2.3.1 Tube leak test: Each tube must be leak tested as soon as it is loaded into the carrier gas flow path before analysis to ensure data integrity.
11.2.3.2 Conduct the leak test at the GC carrier gas pressure, without heat or gas flow applied. Tubes that fail the leak test should not be analyzed, but should be resealed and stored intact. On automated systems, the instrument should continue to leak test and analyze subsequent tubes after a given tube has failed. Automated systems must also store and record which tubes in a sequence have failed the leak test. Information on failed tubes should be downloaded with the batch of sequence information from the analytical system.
11.2.3.3 Leak test the sample flow path. Leak check the sample flow path of the thermal desorber before each analysis without heat or gas flow applied to the sample tube. Stop the automatic sequence of tube desorption and GC analysis if any leak is detected in the main sample flow path. This process may
be carried out as a separate step or as part of Section 11.2.3.2.
11.2.4 Optional dry purge.
11.2.4.1 Tubes may be dry purged with a flow of pure dry gas passing into the tube from the sampling end, to remove water vapor and other very volatile interferents if required.
11.2.5 Internal standard (IS) addition.
11.2.5.1 Use the internal standard addition function of the automated thermal desorber (if available) to introduce a precise aliquot of the internal standard to the sampling end of each tube after the leak test and shortly before primary (tube) desorption).
Note: This step can be combined with dry purging the tube (Section 11.2.4) if required.
11.2.5.2 If the analyzer does not have a facility for automatic IS addition, gas or liquid internal standard can be manually introduced to the sampling end of tubes in a flow of carrier gas using the types of procedure described in Sections
10.3 and 10.4, respectively.
11.2.6 Pre-purge. Each tube should be purged to vent with carrier gas flowing in the desorption direction (i.e., flowing
into the tube from the non-sampling end) to remove oxygen before heat is applied. This is to prevent analyte and sorbent oxidation and to prevent deterioration of key analyzer
components such as the GC column and mass spectrometer (if applicable). A series of schematics illustrating these steps is presented in Figures 17.2 and 17.3.
11.3 Analytical Procedure.
11.3.1 Steps Required for Thermal Desorption.
11.3.1.1 Ensure that the pressure and purity of purge and carrier gases supplying the TD/GC/MS system, meet manufacturer specifications and the requirements of this method.
11.3.1.2 Ensure also that the analytical method selected meets the QC requirements of this method (Section 9) and that all the analytical parameters are at set point.
11.3.1.3 Conduct predesorption system checks (see Section 11.2).
11.3.1.4 Desorb the sorbent tube under conditions demonstrated to achieve >95 percent recovery of target compounds (see Section 9.5.2).
Note: Typical tube desorption conditions range from 280-350
C for 5-15 minutes with a carrier gas flow of 30-100 mL/min passing through the tube from the non-sampling end such that analytes are flushed out of the tube from the sampling end. Desorbed VOCs are concentrated (refocused) on a secondary, cooled sorbent trap integrated into the analytical equipment (see Figure 17.4). The focusing trap is typically maintained at a temperature between -30 and +30 �C during focusing. Selection
of hydrophobic sorbents for focusing and setting a trapping temperature of +25 to 27 �C aid analysis of humid samples because these settings allow selective elimination of any residual water from the system, prior to GC/MS analysis.
Note: The transfer of analytes from the tube to the focusing trap during primary (tube) desorption can be carried out splitless or under controlled split conditions (see Figure 17.4) depending on the masses of target compounds sampled and the requirements of the system—sensitivity, required calibration range, column overload limitations, etc. Instrument controlled sample splits must be demonstrated by showing the reproducibility using calibration standards. Field and laboratory blank samples must be analyzed at the same split as the lowest calibration standard. During secondary (trap) desorption the focusing trap is heated rapidly (typically at rates > 40 �C/s) with inert (carrier) gas flowing through the trap (3-100 mL/min) in the reverse direction to that used during focusing.
11.3.1.5 The split conditions selected for optimum field sample analysis must also be demonstrated on representative standards.
Note: Typical trap desorption temperatures are in the range 250-360 C, with a �hold� time of 1-3 minutes at the highest temperature. Trap desorption automatically triggers the start of
GC analysis. The trap desorption can also be carried out under splitless conditions (i.e., with everything desorbed from the
trap being transferred to the analytical column and GC detector) or, more commonly, under controlled split conditions (see Figure 17.4). The selected split ratio depends on the masses of target compounds sampled and the requirements of the system— sensitivity, required calibration range, column overload limitations, etc. If a split is selected during both primary (trap) desorption and secondary (trap) desorption, the overall split ratio is the product of the two. Such �double� split capability gives optimum flexibility for accommodating concentrated samples as well as trace-level samples on the TD/GC/MS analytical system. High resolution capillary columns and most GC/MS detectors tend to work best with approximately
20-200 ng per compound per tube to avoid saturation. The overall split ratio must be adjusted such that, when it is applied to the sample mass that is expected to be collected during field monitoring, the amount reaching the column will be attenuated to fall within this range. As a rule of thumb this means that ~20 ng samples will require splitless or very low split analysis, ~2 �g samples will require a split ratio in the order of ~50:1 and
200 �g samples will require a double split method with an overall split ratio in the order of 2,000:1.
11.3.1.6 Analyzed tubes must be resealed with long-term
storage caps immediately after analysis (manual systems) or after completion of a sequence (automated systems). This prevents contamination, minimizing the extent of tube reconditioning required before subsequent reuse.
11.3.2 GC/MS Analytical Procedure.
11.3.2.1 Heat/cool the GC oven to its starting set point.
11.3.2.2 If using a GC/MS system, it can be operated in either MS-Scan or MS-SIM mode (depending on required sensitivity levels and the type of mass spectrometer selected). As soon as trap desorption and transfer of analytes into the GC column triggers the start of the GC/MS analysis, collect mass spectral data over a range of masses from 35 to 300 amu. Collect at least
10 data points per eluting chromatographic peak in order to adequately integrate and quantify target compounds.
11.3.2.3 Use secondary ion quantitation only when there are sample matrix interferences with the primary ion. If secondary ion quantitation is performed, flag the data and document the reasons for the alternative quantitation procedure.
11.3.2.4 Data reduction is performed by the instruments post processing program that is automatically accessed after data acquisition is completed at the end of the GC run. The concentration of each target compound is calculated using the previously established response factors for the CCV analyzed in Section 11.1.1.6.
11.3.2.5 Whenever the thermal desorption - GC/MS analytical method is changed or major equipment maintenance is performed, you must conduct a new five-level calibration (see Section 10.6.3). System calibration remains valid as long as results from subsequent CCV are within 30 percent of the most recent 5- point calibration (see Section 10.9.5). Include relevant CCV data in the supporting information in the data report for each set of samples.
11.3.2.6 Document, flag and explain all sample results that exceed the calibration range. Report flags and provide documentation in the analytical results for the affected sample(s).
12.1 Data Analysis, Calculations, and Reporting
12.2 Recordkeeping Procedures for Sorbent Tubes.
12.2.1 Label sample tubes with a unique identification number as described in Section 6.3.
12.2.2 Keep records of the tube numbers and sorbent lots used for each sampling period.
12.2.3 Keep records of sorbent tube packing if tubes are manually prepared in the laboratory and not supplied commercially. These records must include the masses and/or bed lengths of sorbent(s) contained in each tube, the maximum allowable temperature for that tube and the date each tube was packed. If a tube is repacked at any stage, record the date of
tube repacking and any other relevant information required in Section 12.1.
12.2.4 Keep records of the conditioning and blanking of tubes. These records must include, but are not limited to, the unique identification number and measured background resulting from the tube conditioning.
12.2.5 Record the location, dates, tube identification and times associated with each sample collection. Record this information on a Chain of Custody form that is sent to the analytical laboratory.
12.2.6 Field sampling personnel must complete and send a Chain of Custody to the analysis laboratory (see Section 8.6.4 of Method 325A for what information to include and Section 17.0 of this method for an example form). Duplicate copies of the Chain of Custody must be included with the sample report and stored with the field test data archive.
12.2.7 Field sampling personnel must also keep records of the unit vector wind direction, sigma theta, temperature and barometric pressure averages for the sampling period. See Section 8.3.4 of Method 325A.
12.2.8 Laboratory personnel must record the sample receipt date, and analysis date.
12.2.9 Laboratory personnel must maintain records of the analytical method and sample results in electronic or hardcopy
in sufficient detail to reconstruct the calibration, sample, and quality control results from each sampling period.
12.2 Calculations.
12.2.1 Complete the calculations in this section to determine compliance with calibration quality control criteria (see also Table 17.1).
12.2.1.1 Response factor (RF). Calculate the RF using
Equation 12.1:
A M
![]()
RF s is
Ais M s
Eq. 12.1
Where:
As Ais |
= = |
Peak area for the characteristic Peak area for the characteristic |
ion of the analyte. ion of the internal |
standard. |
|||
Ms |
= |
Mass of the analyte. |
|
Mis |
= |
Mass of the internal standard. |
12.2.1.2 Standard deviation of the response factors (SDRF).
Calculate the SDRF using Equation 12.2:
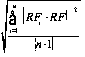 SDRF
SDRF
Eq. 12.2
Where:
RFi = RF for each of the calibration compounds.
RF = Mean RF for each compound from the initial calibration.
n = Number of calibration standards.
12.2.1.3 Percent deviation (%DEV). Calculate the %DEV using Equation 12.3:
%DEV SDRF RF 100
Eq. 12.3
Where:
SDRF = Standard deviation.
RF = Mean RF for each compound from the initial calibration.
12.2.1.4 Relative percent difference (RPD). Calculate the
RPD using Equation 12.4:
RPD
R1 R2
![]() R1 R2/ 2100
R1 R2/ 2100
Eq. 12.4
Where:
R1, R2 = Values that are being compared (i.e., response factors in CCV).
12.2.2 Determine the equivalent concentration of compounds in atmospheres as follows.
12.2.3 Correct target concentrations determined at the sampling site temperature and atmospheric pressure to standard conditions (25C and 760 mm mercury) using Equation 12.5 (Reference 21).
3
![]()
298.2 2
Pss
U NTP
U t
![]() 760
760
Eq. 12.5
![]() ss
ss
Where:
tss = The average temperature during the collection period at the sampling site (K).
Pss = The average pressure at the sampling site during the collection period (mm Hg).
U = The diffusive uptake rate (sampling rate) (mL/min).
12.2.4 For passive sorbent tube samples, calculate the concentration of the target compound(s) in the sampled air, in �g/m3 by using Equation 12.6 (Reference 22).
Cm
mmeas UNTP t
![]() 106
106
Eq. 12.6
Where:
Cm = The concentration of target compound in the air sampled (�g/m3).
mmeas = The mass of the compound as measured in the sorbent tube (�g).
UNTP = The diffusive uptake rate corrected for local conditions (sampling rate) (mL/min).
t = The exposure time (minutes).
Note: Diffusive uptake rates for common VOCs, using carbon sorbents packed into sorbent tubes of the dimensions specified in Section 6.1, are listed in Table 12.1. Adjust analytical conditions to keep expected sampled masses within range (see Sections 11.3.1.3 to 11.3.1.5). Best possible method detection limits are typically in the order of 0.1 ppb for 1,3-butadiene and 0.05 ppb for volatile aromatics such as benzene for 14-day monitoring. However, actual detection limits will depend upon the analytical conditions selected.
Table 12.1: Validated Sorbents and Uptake Rates (mL/min) for Selected Clean Air Act Compounds
Compound |
CarbopackTM Xa |
CarbographTM 1 TD |
CarbopackTM B |
1,1-Dichloroethene |
0.57�0.14 |
not available |
not available |
3-Chloropropene |
0.51�0.3 |
not available |
not available |
1,1-Dichloroethane |
0.57�0.1 |
not available |
not available |
1,2-Dichloroethane |
0.57�0.08 |
not available |
not available |
1,1,1-Trichloroethane |
0.51�0.1 |
not available |
not available |
Benzene |
0.67�0.06 |
0.63�0.07b |
0.63�0.07b |
Carbon tetrachloride |
0.51�0.06 |
not available |
not available |
1,2-Dichloropropane |
0.52�0.1 |
not available |
not available |
Trichloroethene |
0.5�0.05 |
not available |
not available |
1,1,2-Trichloroethane |
0.49�0.13 |
not available |
not available |
Toluene |
0.52�0.14 |
0.56�0.06c |
0.56�0.06c |
Tetrachloroethene |
0.48�0.05 |
not available |
not available |
Chlorobenzene |
0.51�0.06 |
not available |
not available |
Ethylbenzene |
0.46�0.07 |
not available |
0.50c |
m,p-Xylene |
0.46�0.09 |
0.47�0.04c |
0.47�0.04c |
Styrene |
0.5�0.14 |
not available |
not available |
o-Xylene |
0.46�0.12 |
0.47�0.04c |
0.47�0.04c |
p-Dichlorobenzene |
0.45�0.05 |
not available |
not available |
a Reference 3, McClenny, J. Environ. Monit. 7:248-256. Based on 24-hour duration.
b Reference 24, BS EN 14662-4:2005 (incorporated by reference-see �63.14). Based on 14-day duration.
c Reference 25, ISO 16017-2:2003(E) (incorporated by reference-see �63.14). Based on 14-day duration.
13.1 Method Performance
The performance of this procedure for VOC not listed in Table 12.1 is determined using the procedure in Addendum A of this Method or by one of the following national/international standard methods: ISO 16017-2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN 14662-4:2005 (all incorporated by reference-see
�63.14).
13.2 The valid range for measurement of VOC is approximately 0.5 �g/m3 to 5 mg/m3 in air, collected over a 14-
day sampling period. The upper limit of the useful range depends on the split ratio selected (Section 11.3.1) and the dynamic range of the analytical system. The lower limit of the useful range depends on the noise from the analytical instrument detector and on the blank level of target compounds or interfering compounds on the sorbent tube (see Section 13.3).
13.3 Diffusive sorbent tubes compatible with passive sampling and thermal desorption methods have been evaluated at relatively high atmospheric concentrations (i.e., mid-ppb to
ppm) and published for use in workplace air and industrial/mobile source emissions (References 15-16, 21-22).
13.4 Best possible detection limits and maximum quantifiable concentrations of air pollutants range from sub- part-per-trillion (sub-ppt) for halogenated species such as CCl4 and the freons using an electron capture detector (ECD), SIM Mode GC/MS, triple quad MS or GC/TOF MS to sub-ppb for volatile hydrocarbons collected over 72 hours followed by analysis using GC with quadrupole MS operated in the full SCAN mode.
13.3.1 Actual detection limits for atmospheric monitoring vary depending on several key factors. These factors are:
� Minimum artifact levels.
� GC detector selection.
� Time of exposure for passive sorbent tubes.
� Selected analytical conditions, particularly column resolution and split ratio.
14.0 Pollution Prevention
This method involves the use of ambient concentrations of gaseous compounds that post little or no danger of pollution to the environment.
15.0 Waste Management
Dispose of expired calibration solutions as hazardous materials. Exercise standard laboratory environmental practices to minimize the use and disposal of laboratory solvents.
16.1 References
1. Winberry, W. T. Jr., et al., Determination of Volatile Organic Compounds in Ambient Air Using Active Sampling onto Sorbent Tubes: Method TO-17r, Second Edition, U.S. Environmental Protection Agency, Research Triangle Park, NC 27711, January 1999. https://www.epa.gov/ttnamti1/airtox.html#compendium
2. Ciccioli, P., Brancaleoni, E., Cecinato, A., Sparapini, R., and Frattoni, M., "Identification and Determination of Biogenic and Anthropogenic VOCs in Forest Areas of Northern and Southern Europe and a Remote Site of the Himalaya Region by High-resolution GC-MS," J. of Chrom., 643, pp 55-69, 1993.
3. McClenny, W.A., K.D. Oliver, H.H. Jacumin, Jr., E.H. Daughtrey, Jr., D.A. Whitaker. 2005. 24 h diffusive sampling of
toxic VOCs in air onto CarbopackTM X solid adsorbent followed by thermal desorption/GC/MS analysis– laboratory studies. J. Environ. Monit. 7:248-256.
4. Markes International (www.markes.com/publications):
Thermal desorption Technical Support Note 2: Prediction of uptake rates for diffusive tubes.
5. Ciccioli, P., Brancaleoni, E., Cecinato, A., DiPalo, C., Brachetti, A., and Liberti, A., "GC Evaluation of the Organic Components Present in the Atmosphere at Trace Levels with the Aid of Carbopack� B for Preconcentration of the Sample," J. of Chrom., 351, pp 433-449, 1986.
6. Broadway, G. M., and Trewern, T., "Design Considerations for the Optimization of a Packed Thermal Desorption Cold Trap for Capillary Gas Chromatography," Proc. 13th Int�l Symposium on Capil. Chrom., Baltimore, MD, pp 310- 320, 1991.
7. Broadway, G. M., "An Automated System for use Without Liquid Cryogen for the Determination of VOC�s in Ambient Air," Proc. 14th Int�l. Symposium on Capil. Chrom., Baltimore, MD, 1992.
8. Gibitch, J., Ogle, L., and Radenheimer, P., "Analysis of Ozone Precursor Compounds in Houston, Texas Using Automated Continuous GCs," in Proceedings of the Air and Waste Management Association Conference: Measurement of Toxic and Related Air
Pollutants, Air and Waste Management Association, Pittsburgh, PA, May 1995.
9. Vandendriessche, S., and Griepink, B., "The Certification of Benzene, Toluene and m-Xylene Sorbed on Tenax� TA in Tubes," CRM-112 CEC, BCR, EUR12308 EN, 1989.
10. MDHS 2 (Acrylonitrile in Air), "Laboratory Method Using Porous Polymer Adsorption Tubes, and Thermal Desorption with Gas Chromatographic Analysis," Methods for the Determination of Hazardous Substances (MDHS), UK Health and Safety Executive, Sheffield, UK.
11. MDHS 22 (Benzene in Air), "Laboratory Method Using Porous Polymer Adsorbent Tubes, Thermal Desorption and Gas Chromatography," Method for the Determination of Hazardous Substances (MDHS), UK Health and Safety Executive, Sheffield, UK.
12. MDHS 23 (Glycol Ether and Glycol Acetate Vapors in Air), "Laboratory Method Using Tenax� Sorbent Tubes, Thermal Desorption and Gas Chromatography," Method for the Determination of Hazardous Substances (MDHS), UK Health and Safety Executive, Sheffield, UK.
13. MDHS 40 (Toluene in air), "Laboratory Method Using Pumped Porous Polymer Adsorbent Tubes, Thermal Desorption and Gas Chromatography," Method for the Determination of Hazardous Substances (MDHS), UK Health and Safety Executive, Sheffield,
UK.
14. MDHS 60 (Mixed Hydrocarbons (C to C) in Air), "Laboratory Method Using Pumped Porous Polymer 3 10 and Carbon Sorbent Tubes, Thermal Desorption and Gas Chromatography," Method for the Determination of Hazardous Substances (MDHS), UK Health and Safety Executive, Sheffield, UK.
15. Price, J. A., and Saunders, K. J., "Determination of Airborne Methyl tert-Butyl Ether in Gasoline Atmospheres," Analyst, Vol. 109, pp. 829-834, July 1984.
16. Coker, D. T., van den Hoed, N., Saunders, K. J., and Tindle, P. E., "A Monitoring Method for Gasoline Vapour Giving Detailed Composition," Ann. Occup, Hyg., Vol 33, No. 11, pp 15- 26, 1989.
17. DFG, "Analytische Methoden zur prufing gesundheitsschadlicher Arbeistsstoffe," Deutsche Forschungsgemeinschaft, Verlag Chemie, Weinheim FRG, 1985.
18. NNI, "Methods in NVN Series (Luchtkwaliteit; Werkplekatmasfeer)," Nederlands Normailsatie - Institut, Delft, The Netherlands, 1986-88.
19. "Sampling by Solid Adsorption Techniques," Standards Association of Australia Organic Vapours, Australian Standard 2976, 1987.
20. Woolfenden, E. A., "Monitoring VOCs in Air Using Pumped Sampling onto Sorbent Tubes Followed by Thermal
Desorption-capillary GC Analysis: Summary of Reported Data and Practical Guidelines for Successful Application," J. Air & Waste Manage. Assoc., Vol. 47, 1997, pp. 20-36.
21. Validation Guidelines for Air Sampling Methods Utilizing Chromatographic Analysis, OSHA T-005, Version 3.0, May 2010, https://www.osha.gov/dts/sltc/methods/chromguide/chromguide.pdf.
22. ASTM D4597-10, Standard Practice for Sampling Workplace Atmospheres to collect Gases or Vapors with Solid Sorbent Diffusive Samplers.
23. Martin, https://www.hsl.gov.uk/media/1619/issue14.pdf.
24. BS EN 14662-4:2005, Ambient air quality – Standard method for the measurement of benzene concentrations – Part 4: Diffusive sampling followed by thermal desorption and gas chromatography.
25. ISO 16017-2:2003(E): Indoor, ambient and workplace air
– Sampling and analysis of volatile organic compounds by sorbent tube/thermal desorption/capillary gas chromatography – Part 2: Diffusive sampling.
17.0 Tables, Diagrams, Flowcharts and Validation Data
Table 17.1. Summary of GC/MS Analysis Quality Control Procedures
Parameter |
Frequency |
Acceptance Criteria |
Corrective Action |
Bromofluorobenzene Instrument Tune Performance Check |
Dailya prior to sample analysis |
Evaluation criteria presented in |
1) Retune and or 2) Perform Maintenance |
Section 9.5 and Table 9.2. |
|||
Five point calibration bracketing the expected sample concentration. |
Following any major change, repair or maintenance or if daily CCV does not meet method requirements. Recalibration not to exceed three months. |
1) Percent Deviation (%DEV) of response factors �30% 2) Relative Retention Times (RRTs) for target peaks �0.06 units from mean RRT |
1) Repeat calibration sample analysis 2) Repeat linearity check 3) Prepare new calibration standards as necessary and repeat analysis |
Calibration Verification (CCV Second source calibration verification check) |
Following the calibration curve |
The response factor �30% DEV from calibration curve average response factor |
1) Repeat calibration check 2) Repeat calibration curve |
Laboratory Blank |
Dailya following |
1) �0.2 ppbv per |
1) Repeat |
Analysis |
bromofluorobenzen |
analyte or � 3 |
analysis with new |
e and calibration |
times the LOD, |
blank tube |
|
check; prior to sample analysis |
whichever is greater |
2) Check system for leaks, |
|
2) Internal |
contamination |
||
Standard (IS) area response �40% and IS |
3) Analyze additional blank |
||
Retention Time |
|||
(RT) �0.33 min. |
|||
of most recent |
|||
calibration check |
|||
Blank Sorbent Tube |
One tube analyzed |
<0.2 ppbv per VOC |
Reclean all tubes |
Certification |
for each batch of |
targeted compound |
in batch and |
tubes cleaned or |
or 3 times the |
reanalyze |
|
10 percent of |
LOD, whichever is |
||
tubes whichever |
greater |
||
is greater. |
|||
Samples - Internal |
All samples |
IS area response |
Flag Data for |
Standards |
�40% and IS RT |
possible |
|
�0.33 min. of |
invalidation |
||
most recent |
|||
calibration |
|||
validation |
a Every 24 hours
Method 325 A/B
EXAMPLE FIELD TEST DATA SHEET (FTDS) AND
CHAIN OF CUSTODY
I. GENERAL INFORMATION
SITE NAME:
SITE LOCATION ADDRESS:
CITY: STATE: ZIP:
II. SAMPLING DATA
Sample ID (Tube) |
Sample or |
Start |
Start |
Stop |
Stop |
Location |
Ambient Temp. |
Barometric Pressure |
|
# |
Sorbent |
blank |
Date |
Time |
Date |
Time |
(gps) |
(F) |
(in. Hg) |
III. CUSTODY INFORMATION
COLLECTED BY:
Relinquished to Shipper -
Name: Received by Laboratory - Name
Date:
Date:
Time
Time
Sample condition upon receipt:
Analysis Required:
Comments:
Figure 17.1. Example Field Data From and Chain of Custody
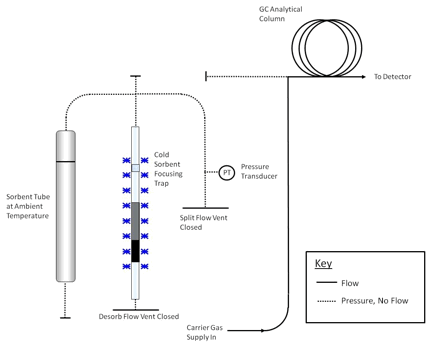
Figure 17.2. Schematic of Thermal Desorption Flow Path During Leak Testing
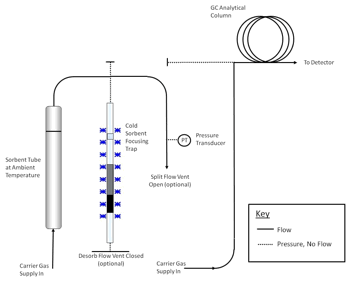
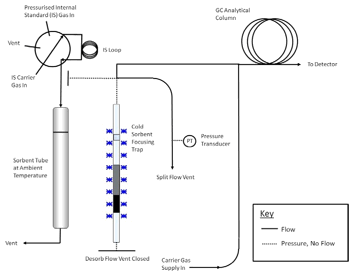
Figure 17.3. Schematic of Thermal Desorption Flow During Purge of Air (Top) and Addition of IS Gas to the Sorbent Tube (Bottom)
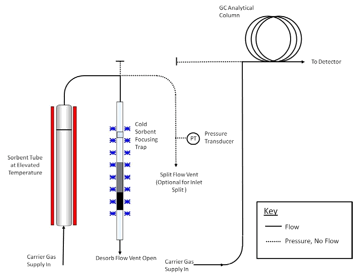
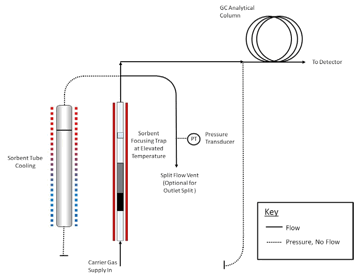
Figure 17.4. Schematic of Thermal Desorption Flow Path During Primary (Tube) Desorption (Top) and Secondary (Trap) Desorption and Transfer to the GC (Bottom)
ADDENDUM A to Method 325B--Method 325 Performance Evaluation
A.1 Scope and Application
A.1.1 To be measured by Methods 325A and 325B, each new target volatile organic compound (VOC) or sorbent that is not listed in Table 12.1 must be evaluated by exposing the selected sorbent tube to a known concentration of the target compound(s) in an exposure chamber following the procedure in this Addendum or by following the procedures in the national/international standard methods: ISO 16017-2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN 14662-4:2005 (all incorporated by reference—see
�63.14), or reported in peer-reviewed open literature.
A.1.2 You must determine the uptake rate and the relative standard deviation compared to the theoretical concentration of volatile material in the exposure chamber for each of the tests required in this method. If data that meet the requirement of this Addendum are available in the peer reviewed open literature for VOCs of interest collected on your passive sorbent tube configuration, then such data may be submitted in lieu of the testing required in this Addendum.
A.1.3 You must expose sorbent tubes in a test chamber to parts per trillion by volume (pptv) and low parts per billion by volume (ppbv) concentrations of VOCs in humid atmospheres to determine the sorbent tube uptake rate and to confirm compound capture and recovery.
A.2 Summary of Method
Note: The technique described here is one approach for determining uptake rates for new sorbent/sorbate pairs. It is equally valid to follow the techniques described in any one of the following national/international standards methods: ISO 16017-2:2003(E), ASTM D6196-03 (Reapproved 2009), or BS EN
14662-4:2005 (all incorporated by reference-see �63.14).
A.2.1 Known concentrations of VOC are metered into an exposure chamber containing sorbent tubes filled with media selected to capture the volatile organic compounds of interest (see Figure A.1 and A.2 for an example of the exposure chamber and sorbent tube retaining rack). VOC are diluted with humid air and the chamber is allowed to equilibrate for 6 hours. Clean passive sampling devices are placed into the chamber and exposed for a measured period of time. The passive uptake rate of the passive sampling devices is determined using the standard and dilution gas flow rates. Chamber concentrations are confirmed with whole gas sample collection and analysis or direct interface volatile organic compound measurement methods.
A.2.2 An exposure chamber and known gas concentrations must be used to challenge and evaluate the collection and recovery of target compounds from the sorbent and tube selected to perform passive measurements of VOC in atmospheres.
A.3 Definitions
A.3.1 cc is cubic centimeter.
A.3.2 ECD is electron capture detector.
A.3.3 FID is flame ionization detector.
A.3.4 LED is light-emitting diode.
A.3.5 MFC is mass flow controller.
A.3.6 MFM is mass flow meter.
A.3.7 min is minute.
A.3.8 ppbv is parts per billion by volume.
A.3.9 ppmv is parts per million by volume.
A.3.10 PSD is passive sampling device.
A.3.11 psig is pounds per square inch gauge.
A.3.12 RH is relative humidity.
A.3.13 VOC is volatile organic compound.
A.4 Interferences
A.4.1 VOC contaminants in water can contribute interference or bias results high. Use only distilled, organic- free water for dilution gas humidification.
A.4.2 Solvents and other VOC-containing liquids can contaminate the exposure chamber. Store and use solvents and other VOC-containing liquids in the exhaust hood when exposure experiments are in progress to prevent the possibility of contamination of VOCs into the chamber through the chamber�s exhaust vent.
Note: Whenever possible, passive sorbent evaluation should
be performed in a VOC free laboratory.
A.4.3 PSDs should be handled by personnel wearing only clean, white cotton or powder free nitrile gloves to prevent contamination of the PSDs with oils from the hands.
A.4.4 This performance evaluation procedure is applicable to only volatile materials that can be measured accurately with direct interface gas chromatography or whole gas sample collection, concentration and analysis. Alternative methods to confirm the concentration of volatile materials in exposure chambers are subject to Administrator approval.
A.5 Safety
A.5.1 This procedure does not address all of the safety concerns associated with its use. It is the responsibility of the user of this standard to establish appropriate field and laboratory safety and health practices and determine the applicability of regulatory limitations prior to use.
A.5.2 Laboratory analysts must exercise appropriate care in working with high-pressure gas cylinders.
A.6 Equipment and Supplies
A.6.1 You must use an exposure chamber of sufficient size to simultaneously expose a minimum of eight sorbent tubes.
A.6.2 Your exposure chamber must not contain VOC that interfere with the compound under evaluation. Chambers made of glass and/or stainless steel have been used successfully for
measurement of known concentration of selected VOC compounds.
A.6.3 The following equipment and supplies are needed:
� Clean, white cotton or nitrile gloves;
� Conditioned passive sampling device tubes and diffusion caps; and
� NIST traceable high resolution digital gas mass flow meters (MFMs) or flow controllers (MFCs).
A.7 Reagents and Standards
A.7.1 You must generate an exposure gas that contains between 35 and 75 percent relative humidity and a concentration of target compound(s) within 2 to 5 times the concentration to be measured in the field.
A.7.2 Target gas concentrations must be generated with certified gas standards and diluted with humid clean air. Dilution to reach the desired concentration must be done with zero grade air or better.
A.7.3 The following reagents and standards are needed:
� Distilled water for the humidification;
� VOC standards mixtures in high-pressure cylinder certified by the supplier (Note: The accuracy of the
certified standards has a direct bearing on the accuracy of the measurement results. Typical vendor accuracy is �5
percent accuracy but some VOC may only be available at lower accuracy (e.g., acrolein at 10 percent)); and
� Purified dilution air containing less than 0.2 ppbv of the target VOC.
A.8 Sample Collection, Preservation and Storage
A.8.1 You must use certified gas standards diluted with humid air. Generate humidified air by adding distilled organic free water to purified or zero grade air. Humidification may be accomplished by quantitative addition of water to the air dilution gas stream in a heated chamber or by passing purified air through a humidifying bubbler. You must control the relative humidity in the test gas throughout the period of passive sampler exposure.
Note: The RH in the exposure chamber is directly proportional to the fraction of the purified air that passes through the water in the bubbler before entering the exposure chamber. Achieving uniform humidification in the proper range is a trial-and-error process with a humidifying bubbler. You may need to heat the bubbler to achieve sufficient humidity. An equilibration period of approximately 15 minutes is required following each adjustment of the air flow through the humidifier. Several adjustments or equilibration cycles may be required to achieve the desired RH level.
Note: You will need to determine both the dilution rate and
the humidification rate for your design of the exposure chamber by trial and error before performing method evaluation tests.
A.8.2 Prepare and condition sorbent tubes following the procedures in Method 325B Section 7.0.
A.8.3 You must verify that the exposure chamber does not
leak.
A.8.4 You must complete two evaluation tests using a minimum of eight passive sampling tubes in each test with less than 5-percent depletion of test analyte by the samplers.
A.8.4.1 Perform at least one evaluation at two to five times the estimated analytical detection limit or less.
A.8.4.2 Perform second evaluation at a concentration equivalent to the middle of the analysis calibration range.
A.8.5 You must evaluate the samplers in the test chamber operating between 35 percent and 75 percent RH, and at 25 �5 �C. Allow the exposure chamber to equilibrate for 6 hours before starting an evaluation.
A.8.6 The flow rate through the chamber must be � 0.5 meter per second face velocity across the sampler face.
A.8.7 Place clean, ready to use sorbent tubes into the exposure chamber for predetermined amounts of time to evaluate collection and recovery from the tubes. The exposure time depends on the concentration of volatile test material in the chamber and the detection limit required for the sorbent tube
sampling application. Exposure time should match sample collection time. The sorbent tube exposure chamber time may not be less than 24 hours and should not be longer than 2 weeks.
A.8.7.1 To start the exposure, place the clean PSDs equipped with diffusion caps on the tube inlet into a retaining rack.
A.8.7.2 Place the entire retaining rack inside the exposure chamber with the diffusive sampling end of the tubes facing into the chamber flow. Seal the chamber and record the exposure start time, chamber RH, chamber temperature, PSD types and numbers, orientation of PSDs, and volatile material mixture composition (see Figure A.2).
A.8.7.3 Diluted, humidified target gas must be continuously fed into the exposure chamber during cartridge exposure. Measure the flow rate of target compound standard gas and dilution air to an accuracy of 5 percent.
A.8.7.4 Record the time, temperature, and RH at the beginning, middle, and end of the exposure time.
A.8.7.5 At the end of the exposure time, remove the PSDs from the exposure chamber. Record the exposure end time, chamber RH, and temperature.

Figure A.1. Example Sorbent Tube Exposure Chamber


Figure A.2. Example Tube Retaining Rack in Exposure Chamber
A.9 Quality Control
A.9.1 Monitor and record the exposure chamber temperature and RH during PSD exposures.
A.9.2 Measure the flow rates of standards and purified
humified air immediately following PSD exposures. A.10 Calibration and Standardization
A.10.1 Follow the procedures described in Method 325B Section 10.0 for calibration.
A.10.2 Verify chamber concentration by direct injection into a gas chromatograph calibrated for the target compound(s) or by collection of an integrated SUMMA canister followed by analysis using a preconcentration gas chromatographic method such as EPA Compendium Method TO–15, Determination of VOCs in Air Collected in Specially-Prepared Canisters and Analyzed By GC/MS.
A.10.2.1 To use direct injection gas chromatography to verify the exposure chamber concentration, follow the procedures in Method 18 of 40 CFR part 60, Appendix A-6. The method ASTM D6420-99 (Reapproved 2010) (incorporated by reference—see
�63.14) is an acceptable alternative to EPA Method 18 of 40 CFR part 60).
Note: Direct injection gas chromatography may not be sufficiently sensitive for all compounds. Therefore, the whole gas preconcentration sample and analysis method may be required to measure at low concentrations.
A.10.2.2 To verify exposure chamber concentrations using SUMMA canisters, prepare clean canister(s) and measure the concentration of VOC collected in an integrated SUMMA canister
over the period used for the evaluation (minimum 24 hours). Analyze the TO-15 canister sample following EPA Compendium Method TO-15.
A.10.2.3 Compare the theoretical concentration of volatile material added to the test chamber to the measured concentration to confirm the chamber operation. Theoretical concentration must agree with the measured concentration within 30 percent.
A.11 Analysis Procedure
Analyze the sorbent tubes following the procedures described in Section 11.0 of Method 325B.
A.12 Recordkeeping Procedures for Sorbent Tube Evaluation
Keep records for the sorbent tube evaluation to include at a minimum the following information:
A.12.1 Sorbent tube description and specifications.
A.12.2 Sorbent material description and specifications.
A.12.3 Volatile analytes used in the sampler test.
A.12.4 Chamber conditions including flow rate, temperature, and relative humidity.
A.12.5 Relative standard deviation of the sampler results at the conditions tested.
A.12.6 95 percent confidence limit on the sampler overall accuracy.
A.12.7 The relative accuracy of the sorbent tube results compared to the direct chamber measurement by direct gas
chromatography or SUMMA canister analysis. A.13 Method Performance
A.13.1 Sorbent tube performance is acceptable if the relative accuracy of the passive sorbent sampler agrees with the active measurement method by �10 percent at the 95 percent confidence limit and the uptake ratio is equal to greater than
0.5 mL/min (1 ng/ppm-min).
Note: For example, there is a maximum deviation comparing Perkin-Elmer passive type sorbent tubes packed with CarbopackTM X of 1.3 to 10 percent compared to active sampling using the following uptake rates.
1,3-butadiene uptake rate mL/min |
Estimated Detection Limit (2 week) |
Benzene uptake rates mL/min |
Estimated Detection Limit (2 week) |
|
CarbopackTM X (2 week) |
0.61�0.11a |
0.1 ppbv |
0.67a |
0.05 ppbv |
a McClenny, W.A., K.D. Oliver, H.H. Jacumin, Jr., E.H. Daughtrey, Jr., D.A. Whitaker. 2005. 24 h diffusive sampling of toxic VOCs in air onto CarbopackTM X solid adsorbent followed by thermal desorption/GC/MS analysis– laboratory studies. J. Environ. Monit. 7:248-256.
A.13.2 Data Analysis and Calculations for Method Evaluation
A.13.2.1 Calculate the theoretical concentration of VOC standards using Equation A.1.
C f
![]()
FR
FRi
Cs t FRa
Eq. A.1
Where:
Cf = The final concentration of standard in the exposure chamber (ppbv).
FRi = The flow rate of the target compound I (mL/min).
FRt = The flow rate of all target compounds from separate if multiple cylinders are used (mL/min).
FRa = The flow rate of dilution air plus moisture (mL/min).
Cs = The concentration of target compound in the standard cylinder (parts per million by volume).
A.13.2.3 Determine the uptake rate of the target gas being
evaluated using Equation A.2.
U
Mx
Ce Tt
Eq. A.2
Where:
Mx = The mass of analyte measured on the sampling tube (g). Ce = The theoretical exposure chamber concentration (g/mL). Tt = The exposure time (minutes).
A.13.2.4 Estimate the variance (relative standard deviation (RSD)) of the inter-sampler results at each condition tested using Equation A.3. RSD for the sampler is estimated by pooling the variance estimates from each test run.
X X
![]() n 2
n 2
S 2 i
i n 1
Eq. A.3
Where:
Xi = The measured mass of analyte found on sorbent tube i.
![]() X i =
The mean value of all Xi.
X i =
The mean value of all Xi.
n = The number of measurements of the analyte.
A.13.2.4 Determine the percent relative standard deviation of the inter-sampler results using Equation A.4.
%RSDx
100
![]()
![]() X
X
Eq. A.4
A.13.2.5 Determine the 95 percent confidence interval for the sampler results using Equation A.5. The confidence interval is determined based on the number of test runs performed to evaluate the sorbent tube and sorbent combination. For the minimum test requirement of eight samplers tested at two concentrations, the number of tests is 16 and the degrees of
freedom are 15.
95%
%RSD t0.95 f
Eq. A.5
![]() Where:
Where:
95%
= 95 percent confidence interval.
%RSD = percent relative standard deviation.
t0.95 = The Students t statistic for f degrees of freedom at 95 percent confidence.
f = The number of degrees of freedom. n = Number of samples.
A.13.2.6 Determine the relative accuracy of the sorbent tube combination compared to the active sampling results using Equation A.6.
RA
Xi X A 95%
Eq. A.6
Where:
RA = Relative accuracy.
![]() X i = The mean value of all Xi.
X i = The mean value of all Xi.
X A
95%
= The average concentration of analyte measured by the active measurement method.
= 95 percent confidence interval.
A.14 Pollution Prevention
This method involves the use of ambient concentrations of gaseous compounds that post little or no pollution to the environment.
A.15 Waste Management
Expired calibration solutions should be disposed of as hazardous materials.
A.16 References
1. ISO TC 146/SC 02 N 361 Workplace atmospheres – Protocol for evaluating the performance of diffusive samplers.
![]()
![]()
![]()
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources

Terms of Use
Clean Air Engineering, Inc.
© 2009-2019 Clean Air Engineering,
All Rights Reserved
![]()

![]()
Private
Parking Survey