- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 30A--Determination Of Total Vapor Phase Mercury Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What is Method 30A?
Method 30A is a procedure for measuring total vapor phase mercury (Hg) emissions from stationary sources using an instrumental analyzer. This method is particularly appropriate for performing emissions testing and for conducting relative accuracy test audits (RATAs) of mercury continuous emissions monitoring systems (Hg CEMS) and sorbent trap monitoring systems at coal-fired combustion sources. Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known and acceptable quality for each testing site. This method does not completely describe all equipment, supplies, and sampling procedures and analytical procedures you will need but refers to other test methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional methods which are also found in appendices A-1 and A-3 to this part:
(a) Method 1—Sample and Velocity Traverses for Stationary Sources.
(b) Method 4—Determination of Moisture Content in Stack Gases.
1.1 Analytes.
What does this method determine? This method is designed to measure the mass concentration of total vapor phase Hg in flue gas, which represents the sum of elemental Hg (Hg0) and oxidized forms of Hg (Hg+2), in mass concentration units of micrograms per cubic meter (μg/m3).
| Analyte | CAS No. | Sensitivity |
| Elemental Hg (Hg0) | 7439-97-6 | Typically <2% of calibration Span |
| Oxidized Hg (Hg+2) | ----- | (Same) |
1.2 Applicability.
When is this method required? Method 30A is offered as a reference method for emission testing and for RATAs of Hg CEMS and sorbent trap monitoring systems at coal-fired boilers. Method 30A may also be specified for other source categories in the future, either by New Source Performance Standards (NSPS), National Emission Standards for Hazardous Air Pollutants (NESHAP), emissions trading programs, State Implementation Plans (SIP), or operating permits that require measurement of Hg concentrations in stationary source emissions to determine compliance with an applicable emission standard or limit, or to conduct RATAs of Hg CEMS and sorbent trap monitoring systems.
1.3 Data Quality Objectives (DQO).
How good must my collected data be? Method 30A has been designed to provide data of high and known quality for Hg emission testing and for relative accuracy testing of Hg monitoring systems including Hg CEMS and sorbent trap monitoring systems. In these and other applications, the principle objective is to ensure the accuracy of the data at the actual emission levels encountered. To meet this objective, calibration standards prepared according to an EPA traceability protocol must be used and measurement system performance tests are required.
2.0 Summary of Method
In this method, a sample of the effluent gas is continuously extracted and conveyed to an analyzer capable of measuring the total vapor phase Hg concentration. Elemental and oxidized mercury (i.e., Hg0 and Hg+2) may be measured separately or simultaneously but, for purposes of this method, total vapor phase Hg is the sum of Hg0 and Hg+2. You must meet the performance requirements of this method (i.e.., system calibration, interference testing, dynamic spiking, and system integrity/drift checks) to validate your data. The dynamic spiking requirement is deferred until January 1, 2009.
3.0 Definitions
3.1 calibration Curve means the relationship between an analyzer’s response to the injection of a series of calibration gases and the actual concentrations of those gases.
3.2 calibration Gas means a gas standard containing Hg0 or HgCl2 at a known concentration that is produced and certified in accordance with an EPA traceability protocol for certification of Hg calibration standards.
3.2.1 Zero Gas means a calibration gas with a concentration that is below the level detectable by the measurement system.
3.2.2 Low-Level Gas means a calibration gas with a concentration that is 10 to 30 percent of the calibration span.
3.2.3 Mid-Level Gas means a calibration gas with a concentration that is 40 to 60 percent of the calibration span.
3.2.4 High-Level Gas means a calibration gas whose concentration is equal to the calibration span.
3.3 Converter means a device that reduces oxidized mercury (Hg+2) to elemental mercury (Hg0).
3.4 calibration Span means the upper limit of valid instrument response during sampling. To the extent practicable the measured emissions are to be between 10 and 100 percent of the selected calibration span (i.e., the measured emissions should be within the calibrated range determined by the Low- and High-Level gas standards). It is recommended that the calibration span be at least twice the native concentration to accommodate the dynamic spiking procedure.
3.5 Centroidal Area means the central area that has the same shape as the stack or duct cross section and is no greater than one percent of the stack or duct total cross- sectional area.
3.6 Data Recorder means the equipment that permanently records the concentrations reported by the analyzer.
3.7 Drift Check means the test to determine the difference between the measurement system readings obtained in a post-run system integrity check and the prior pre-run system integrity check at a specific calibration gas concentration level (i.e., zero, mid-level, or high-level).
3.8 Dynamic Spiking means a procedure in which a known mass or concentration of vapor phase HgCl2 is injected into the Probe sample gas stream at a known flow rate, in order to assess the effects of the flue gas matrix on the accuracy of the measurement system.
3.9 gas analyzer means the equipment that detects the total vapor phase Hg being measured and generates an output proportional to its concentration.
3.10 Interference Test means the test to detect analyzer responses to compounds other than Hg, usually gases present in the measured gas stream, that are not adequately accounted for in the calibration procedure and may cause measurement bias.
3.11 Measurement System means all of the equipment used to determine the Hg concentration. The measurement system may generally include the following major subsystems: sample acquisition, Hg+2 to Hg0 converter, sample transport, sample conditioning, flow control/gas manifold, gas analyzer, and data recorder.
3.12 Native Concentration means the total vapor phase Hg concentration in the effluent gas stream.
3.13 NIST means the National Institute of Standards and Technology, located in Gaithersburg, Maryland.
3.14 Response Time means the time it takes for the measurement system, while operating normally at its target sample flow rate or dilution ratio, to respond to a known step change in gas concentration (from a low-level to a high-level gas) and to read within 5 percent of the stable high-level gas response.
3.15 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.16 System calibration Error means the difference between the measured concentration of a low-, mid-, or high-level Hg0 calibration gas and the certified concentration of the gas when it is introduced in system calibration mode.
3.17 System calibration Mode means introducing the calibration gases into the measurement system at the Probe, upstream of all sample conditioning components.
3.18 Test refers to the series of runs required by the applicable regulation.
4.0 Interferences
Interferences will vary among instruments and potential instrument-specific spectral and matrix interferences must be evaluated through the interference test and the dynamic spiking tests.
5.0 Safety
What safety measures should I consider when using this method?
This method may require you to work with hazardous materials and in hazardous conditions. You are encouraged to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user’s manual. Occupational Safety and Health Administration (OSHA) regulations concerning use of compressed gas cylinders and noxious gases may apply.
6.0 Equipment and Supplies.
6.1 What do I need for the measurement system? This method is intended to be applicable to multiple instrumental technologies. You may use any equipment and supplies that meet the following specifications.
6.1.1 All wetted sampling system components, including Probe components prior to the point at which the calibration gas is introduced, must be chemically inert to all Hg species. Materials such as perfluoroalkoxy (PFA) TeflonTM, quartz, treated stainless steel (SS) are examples of such materials. [Note: These materials of construction are required because components prior to the calibration gas injection point are not included in the system calibration error, system integrity, and interference tests.]
6.1.2 The interference, system calibration error, system integrity, drift and dynamic spiking test criteria must all be met by the system used.
6.1.3 The system must be capable of measuring and controlling sample flow rate.
6.1.4 All system components prior to the Hg+2 to Hg0 converter must be maintained at a sample temperature above the acid gas dew point.
6.2 Measurement System Components.
Figure 30A-1 in Section 17.0 is an example schematic of a Method 30A measurement system.
6.2.1 Sample Probe. The Probe must be made of the appropriate materials as noted in Section 6.1.1, heated when necessary (see Section 6.1.4), configured with ports for introduction of calibration and spiking gases, and of sufficient length to traverse all of the sample points.
6.2.2 filter or Other Particulate Removal Device. The filter or other particulate removal device is considered to be a part of the measurement system, must be made of appropriate materials as noted in Section 6.1.1, and must be included in all system tests.
6.2.3 Sample Line. The sample line that connects the Probe to the converter, conditioning system and analyzer must be made of appropriate materials as noted in Section 6.1.1.
6.2.4 Conditioning equipment. For dry basis measurements, a condenser, dryer or other suitable device is required to remove moisture continuously from the sample gas. Any equipment needed to heat the Probe, or sample line to avoid condensation prior to the moisture removal component is also required. For wet basis systems, you must keep the sample above its dew point either by: (1) heating the sample line and all sample transport components up to the inlet of the analyzer (and, for hot-wet extractive systems, also heating the analyzer) or (2) by diluting the sample prior to analysis using a dilution Probe system. The components required to do either of the above are considered to be conditioning equipment.
6.2.5 Sampling pump. A pump is needed to push or pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. If a mechanical sample pump is used and its surfaces are in contact with the sample gas prior to detection, the pump must be leak free and must be constructed of a material that is non-reactive to the gas being sampled (see Section 6.1.1). For dilution- type measurement systems, an ejector pump (eductor) may be used to create a sufficient vacuum that sample gas will be drawn through a critical orifice at a constant rate. The ejector pump may be constructed of any material that is non-reactive to the gas being sampled.
6.2.6 calibration Gas System(s). One or more systems may be needed to introduce calibration gases into the measurement system. A system should be able to flood the sampling Probe sufficiently to prevent entry of gas from the effluent stream.
6.2.7 Dynamic Spiking Port. For the purposes of the dynamic spiking procedure described in Section 8.2.7, the measurement system must be equipped with a port to allow introduction of the dynamic spike gas stream with the sample gas stream, at a point as close as possible to the inlet of the Probe so as to ensure adequate mixing. The same port used for system calibrations and calibration error checks may be used for dynamic spiking purposes.
6.2.8 Sample Gas Delivery. The sample line may feed directly to a converter, to a by-pass valve (for speciating systems), or to a sample manifold. All valve and/or manifold components must be made of material that is non-reactive to the gas sampled and the calibration gas, and must be configured to safely discharge any excess gas.
6.2.9 Hg Analyzer. An instrument is required that continuously measures the total vapor phase Hg in the gas stream and meets the applicable specifications in Section 13.0.
6.2.10 Data Recorder. A recorder, such as a computerized data acquisition and handling system (DAHS), digital recorder, strip chart, or data logger, is required for recording measurement data.
6.3 Moisture Measurement System.
If correction of the measured Hg emissions for moisture is required (see Section 8.5), either Method 4 in appendix A-3 to this part or other moisture measurement methods approved by the Administrator will be needed to measure stack gas moisture content.
7.0 Reagents and Standards
7.1 calibration Gases.
What calibration gases do I need? You will need calibration gases of known concentrations of Hg0 and HgCl2. Special reagents and equipment may be required to prepare the HgCl2 gas standards (e.g., a NIST-traceable solution of HgCl2 and a gas generator equipped with mass flow controllers). The following calibration gas concentrations are required:
7.1.1 High-Level Gas. Equal to the selected calibration span.
7.1.2 Mid-Level Gas. 40 to 60 percent of the calibration span.
7.1.3 Low-Level Gas. 10 to 30 percent of the calibration span.
7.1.4 Zero Gas. No detectable Hg.
7.1.5 Dynamic Spike Gas. The exact concentration of the HgCl2 calibration gas used to perform the pre-test dynamic spiking procedure described in Section 8.2.7 depends on the native Hg concentration in the stack The spike gas must produce a spiked sample concentration above the native concentration, as specified in Section 8.2.7.2.2.
7.2 Interference Test.
What reagents do I need for the interference test? Use 9
the appropriate test gases listed in Table 30A-3 in Section 17.0 (i.e., the potential interferents for the source to be tested, as identified by the instrument manufacturer) to conduct the interference check. These gases need not be of protocol gas quality.
8.0 Sample Collection
Emission Test Procedure
Figure 30A-2 in Section 17.0 presents an overview of the test procedures required by this method. Since you may choose different options to comply with certain performance criteria, you must identify the specific options and associated frequencies you select and document your results in regard to the performance criteria.
8.1 Sample Point Selection.
What sampling site and sampling points do I select?
8.1.1 When this method is used solely for Hg emission testing (e.g., to determine compliance with an emission standard or limit), use twelve sampling points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, you may conduct a stratification test as described in Section 8.1.3 to determine the number and location of the sampling points.
8.1.2 When this method is used for relative accuracy testing of a Hg CEMS or sorbent trap monitoring system, follow the sampling site selection and sampling point layout procedures for gas monitor RATA testing described in the appropriate performance specification or applicable regulation (e.g., Performance Specification 2, section 8.1.3 of appendix B to this part or section 6.5.6 of appendix A to part 75 of this chapter), with one exception. If you elect to perform stratification testing as part of the sampling point selection process, perform the testing in accordance with Section 8.1.3 of this method (see also “Summary Table of QA/QC Requirements” in Section 9.0).
8.1.3 Determination of Stratification.
If you elect to perform stratification testing as part of the sampling point selection process and the test results show your effluent gas stream to be unstratified or minimally stratified, you may be allowed to sample at fewer points or at different points than would otherwise be required.
8.1.3.1 Test Procedure.
To test for stratification, use a Probe of appropriate length to measure the total vapor phase Hg concentration at twelve traverse points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, for a sampling location where stratification is expected (e.g., after a wet scrubber or at a point where dissimilar gas streams are combined together), if a 12-point Hg stratification test has been previously performed at that location and the results of the test showed the location to be minimally stratified or unstratified according to the criteria in section 8.1.3.2, you may perform an abbreviated 3-point or 6-point Hg stratification test at the points specified in section 6.5.6.2(a) of appendix A to part 75 of this chapter in lieu of performing the 12-point test. Sample for a minimum of twice the system response time (see Section 8.2.6) at each traverse point. Calculate the individual point and mean Hg concentrations.
8.1.3.2 Acceptance Criteria and Sampling Point Location.
8.1.3.2.1 If the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a) ± 5 percent of the mean concentration; or (b) ± 0.2 μg/m3 (whichever is less restrictive), the gas stream is considered to be unstratified and you may collect samples from a single point that most closely matches the mean.
8.1.3.2.2 If the 5 percent or 0.2 μg/m3 criterion in Section 8.1.3.2.1 is not met, but the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a) ± 10 percent of the mean; or (b) ± 0.5 μg/m3 (whichever is less restrictive), the gas stream is considered to be minimally stratified, and you may take samples from three points, provided the points are located on the measurement line exhibiting the highest average Hg concentration during the stratification test. If the stack diameter (or equivalent diameter, for a rectangular stack or duct) is greater than 2.4 meters (7.8 ft), locate the three sampling points at 0.4, 1.0, and 2.0 meters from the stack or duct wall. Alternatively, if a RATA required by part 75 of this chapter is being conducted, you may locate the three points at 4.4, 14.6, and 29.6 percent of the duct diameter, in accordance with Method 1 in appendix A-1 to this part. For stack or duct diameters of 2.4 meters (7.8 ft) or less, locate the three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line.
8.1.3.2.3 If the gas stream is found to be stratified because the 10 percent or 0.5 μg/m3 criterion in Section 8.1.3.2.2 is not met, then either locate three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line that exhibited the highest average Hg concentration during the stratification test, or locate twelve traverse points for the test in accordance with Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part; or, if a RATA required by part 75 of this chapter is being conducted, locate six Method 1 points along the measurement line that exhibited the highest average Hg concentration.
8.1.3.3 Temporal Variations.
Temporal variations in the source Hg concentration during a stratification test may complicate the determination of stratification. If temporal variations are a concern, you may use the following procedure to normalize the stratification test data. A second Hg measurement system, i.e., either an installed Hg CEMS or another Method 30A system, is required to perform this procedure. Position the sampling Probe of the second Hg measurement system at a fixed point in the stack or duct, at least one meter from the stack or duct wall. Then, each time that the Hg concentration is measured at one of the stratification test points, make a concurrent measurement of Hg concentration at the fixed point. Normalize the Hg concentration measured at each traverse point, by multiplying it by the ratio of CF,avg to CF, where CF is the corresponding fixed-point Hg concentration measurement, and CF,avg is the average of all of the fixed-point measurements over the duration of the stratification test. Evaluate the results of the stratification test according to section 8.1.3.2, using the normalized Hg concentrations.
8.1.3.4 Stratification Testing Exemption.
Stratification testing need not be performed at a test location where it would otherwise be required to justify using fewer sample points or different sample points, if the owner or operator documents that the Hg concentration in the stack gas is expected to be 3 μg/m3 or less at the time of a Hg monitoring system RATA or an Hg emissions test. To demonstrate that a particular test location qualifies for the stratification testing exemption, representative Hg emissions data must be collected just prior to the RATA or emissions test. At least one hour of Hg concentration data is required for the demonstration. The data used for the demonstration shall be recorded at process operating conditions that closely approximate the operating conditions that will exist during the RATA or emissions test. It is recommended that collection of the demonstration data be integrated with the on-site pretest procedures required by the reference method being used for the RATA or emissions test (whether this method or another approved Hg reference method is used). Quality-assured data from an installed Hg monitoring system may also be used for the demonstration. If a particular test location qualifies for the stratification testing exemption, sampling shall be performed at three points, as described in section 8.1.3.2.2 of this method. The owner or operator shall fully document the method used to collect the demonstration data and shall keep this documentation on file with the data from the associated RATA or Hg emissions test.
8.1.3.5 Interim Alternative Stratification Test Procedures.
In the time period between the effective date of this method and January 1, 2009, you may follow one of the following two procedures. Substitute a stratification test for sulfur dioxide (SO2) for the Hg stratification test described in section 8.1.3.1. If this option is chosen, follow the test procedures in section 6.5.6.1 of appendix A to part 75 of this chapter. Evaluate the test results and determine the sampling point locations according to section 6.5.6.3 of appendix A to part 75 of this chapter. If the sampling location is found to be minimally stratified or unstratified for SO2, it shall be considered minimally stratified or unstratified for Hg. Alternatively, you may forgo stratification testing, assume the gas stream is minimally stratified, and sample at three points as described in section 8.1.3.2.2 of this method.
8.2 Initial Measurement System Performance Tests.
What initial performance criteria must my system meet before I begin sampling? Before measuring emissions, perform the following procedures:
(a) Interference Test;
(b) calibration Gas Verification;
(c) Measurement System Preparation;
(d) 3-Point System calibration Error Test;
(e) System Integrity Check;
(f) Measurement System Response Time Test; and
(g) Dynamic Spiking Test.
8.2.1 Interference Test (Optional).
Your measurement system should be free of known interferences. It is recommended that you conduct this interference test of your measurement system prior to its initial use in the field to verify that the candidate test instrument is free from inherent biases or interferences resulting from common combustion emission constituents. If you have multiple measurement systems with components of the same make and model numbers, you need only perform this interference check on one system and you may also rely on an interference test conducted by the manufacturer on a system having components of the same make and model(s) of the system that you use. The interference test procedure is found in Section 8.6 of this method.
8.2.2 calibration Gas Verification.
How must I verify the concentrations of my calibration gases?
8.2.2.1 Cylinder Gas Standards. When cylinder gas standards are used for Hg0, obtain a certificate from the gas manufacturer and confirm that the documentation includes all information required by an EPA traceability protocol (see Section 16). Confirm that the manufacturer certification is complete and current. Ensure that the calibration gas certifications have not expired.
8.2.2.2 Other calibration Standards. All other calibration standards for HgCl2 and Hg0, such as gas generators, must meet the requirements of an EPA traceability protocol (see Section 16), and the certification procedures must be fully documented in the test report.
8.2.2.3 calibration Span. Select the calibration span (i.e., high-level gas concentration) so that the measured source emissions are 10 to 100 percent of the calibration span. This requirement is waived for applications in which the Hg concentrations are consistently below 1 μg/m3; however, the calibration span for these low-concentration applications shall not exceed 5 μg/m3.
8.2.3 Measurement System Preparation.
How do I prepare my measurement system for use? Assemble, prepare, and precondition the measurement system according to your standard operating procedure. Adjust the system to achieve the correct sampling rate or dilution ratio (as applicable). Then, conduct a 3-point system calibration error test using Hg0 as described in Section 8.2.4, an initial system integrity check using HgCl2 and a zero gas as described in Section 8.2.5, and a pre-test dynamic spiking test as described in Section 8.2.7.
8.2.4 System calibration Error Test.
Conduct a 3-point system calibration error test before the first test run. Use Hg0 standards for this test. Introduce the low-, mid-, and high-level calibration gases in any order, in system calibration mode, unless you desire to determine the system response time during this test, in which case, inject the gases such that the high-level injection directly follows the low-level injection. For non-dilution systems, you may adjust the system to maintain the correct flow rate at the analyzer during the test, but you may not make adjustments for any other purpose. For dilution systems, you must operate the measurement system at the appropriate dilution ratio during all system calibration error checks, and you may make only the adjustments necessary to maintain the proper ratio. After each gas injection, wait until a stable response has been obtained. Record the analyzer’s final, stable response to each calibration gas on a form similar to Table 30A-1 in Section 17.0. For each calibration gas, calculate the system calibration error using Equation 30A-1 in Section 12.2. The calibration error specification in Section 13.1 must be met for the low-, mid-, and high- level gases. If the calibration error specification is not met for all three gases, take corrective action and repeat the test until an acceptable 3-point calibration is achieved.
8.2.5 System Integrity Check.
Perform a two-point system integrity check before the first test run. Use the zero gas and either the mid- or high-level HgCl2 calibration gas for the check, whichever one best represents the total vapor phase Hg concentration levels in the stack. Record the data on a form similar to Table 30A-2 in Section 17.0. The system integrity check specification in Section 13.2 must be met for both the zero gas and the mid- or high-level gas. If the system integrity specification is not met for both gases, take corrective action and repeat the test until an acceptable system integrity check is achieved.
8.2.6 Measurement System Response Time.
The measurement system response time is used to determine the minimum sampling time for each sampling point and is equal to the time that is required for the measured Hg concentration to increase from the stable low-level calibration gas response to a value within 5 percent of the stable high- level calibration gas response during the system calibration error test in Section 8.2.4. Round off the measured system response time to the nearest minute.
8.2.7 Dynamic Spiking Test.
You must perform dynamic spiking prior to the first test run to validate your test data. The purpose of this procedure is to demonstrate that the site-specific flue gas matrix does not adversely affect the accuracy of the measurement system. The specifications in Section 13.5 must be met to validate your data. If these specifications are not met for the pre-test dynamic spiking, you may not proceed with the test until satisfactory results are obtained. For the time period between the effective date of this method and January 1, 2009, the dynamic spiking requirement is waived.
8.2.7.1 How do I perform dynamic spiking? Dynamic spiking is a gas phase application of the method of standard additions, which involves injecting a known quantity of Hg into the measurement system upstream of all sample conditioning components, similar to system calibration mode, except the Probe is not flooded and the resulting sample stream includes both effluent gas and the spike gas. You must follow a written procedure that details how the spike is added to the system, how the spike dilution factor (DF) is measured, and how the Hg concentration data are collected and processed.
8.2.7.2 Spiking Procedure Requirements.
8.2.7.2.1 Spiking Gas Requirements. The spike gas must also be a HgCl2 calibration gas certified by an EPA traceability protocol. You must choose concentrations that can produce the target levels while being injected at a volumetric flow rate that is ≤ 20 percent of the total volumetric flow rate through the measurement system (i.e., sample flow rate plus spike gas flow rate).
8.2.7.2.2 Target Spiking Level. The target level for spiking must be 150 to 200 percent of the native Hg concentration; however, if the native Hg concentration is < 1 μg/m3, set the target level to add between 1 and 4 μg/m3 Hg+2 to the native concentration.
Use Equation 30A-5 in Section 12.5 to calculate the acceptable range of spike gas concentrations at the target level. Then select a spike gas concentration in that range.
8.2.7.2.3 Spike Injections. You must inject spikes in such a manner that the spiking does not alter the total volumetric sample system flow rate and dilution ratio (if applicable). You must collect at least 3 data points, and the relative standard deviation (RSD) specification in Section 13.5 must be met. Each data point represents a single spike injection, and pre- and post-injection measurements of the native Hg concentration (or diluted native concentration, as applicable) are required for each spike injection.
8.2.7.2.4 Spike Dilution Factor (DF). For each spike injection, DF, the dilution factor must be determined. DF is the ratio of the total volumetric flow rate of gas through the measurement system to the spike gas flow rate. This factor must be > 5. The spiking mass balance calculation is directly dependent on the accuracy of the DF determination. As a result, high accuracy total volumetric flow rate and spike gas flowrate measurements are required. These flow rates may be determined by direct or indirect measurement. Calibrated flow meters, venturies, orifices or tracer gas measurements are examples of potential flow measurement techniques.
8.2.7.2.5 Concentrations. The measurement system must record total vapor phase Hg concentrations continuously during the dynamic spiking procedure. It is possible that dynamic spiking at a level close to 200 percent of the native Hg concentration may cause the measured Hg concentration to exceed the calibration span value. Avoid this by choosing a lower spiking level or by recalibration at a higher span. The measurements shall not exceed 120 percent of the calibration span. The “baseline” measurements made between spikes may represent the native Hg concentration (if spike gas flow is stopped between injections) or the native Hg concentration diluted by blank or carrier gas flowing at the same rate as the spike gas (if gas flow cannot be stopped between injections). Each baseline measurement must include at least 4 readings or 1 minute (whichever is greater) of stable responses. Use Equation 30A-10 or 30A-11 in Section 12.10 (as applicable) to convert baseline measurements to native concentration.
8.2.7.2.6 Recovery. Calculate spike recoveries using Equation 30A-7 in Section 12.7. Mass recoveries may be calculated from stable responses based on injected mass flows or from integrated response peaks based on total mass injected. Calculate the mean and RSD for the three (or more) spike injections and compare to the specifications in Section 13.5.
8.2.7.2.7 Error Adjustment Option. You may adjust the measurement data collected during dynamic spiking for the system calibration error using Equation 30A-3 in Section 12. To do this, perform the initial system integrity check prior to the dynamic spiking test, and perform another system integrity check following the dynamic spiking test and before the first test run. If you choose this option, you must apply Equation 30A- 3 to both the spiked sample concentration measurement (Css) and the baseline or native concentration measurement (Cnative), each substituted in place of Cavg in the equation.
8.2.7.3 Example Spiking Procedure Using a Hot Vapor calibration Source Generator.
(a) Introduce the spike gas into the Probe using a hot vapor calibration source generator and a solution of HgCl2 in dilute HCl and HNO3. The calibrator uses a mass flow controller (accurate within 2 percent) to measure the gas flow, and the solution feed is measured using a top-loading balance accurate to 0.01g. The challenges of injecting oxidized Hg may make it impractical to stop the flow of gas between spike injections. In this case, operate the hot vapor calibration source generator continuously during the spiking procedure, swapping blank solutions for HgCl2 solutions when switching between spiking and baseline measurements.
(b) If applicable, monitor the measurement system to make sure the total sampling system flow rate and the sample dilution ratio do not change during this procedure. Record all data on a data sheet similar to Table 30A-5 in Section 17.0. If the Hg measurement system design makes it impractical to measure the total volumetric flow rate through the system, use a spike gas that includes a tracer for measuring the dilution factor, DF (see Equation 30A-9 in Section 12.9). Allow the measurements to stabilize between each spike injection, average the pre- and post-injection baseline measurements, and calculate the native concentration. If this measurement shifts by more than 5 percent during any injection, it may be necessary to discard that data point and repeat the injection to achieve the required RSD among the injections. If the spikes persistently show poor repeatability, or if the recoveries are not within the range specified in Section 13.5, take corrective action.
8.2.8 Run Validation.
How do I confirm that each run I conduct is valid ?
8.2.8.1 System Integrity Checks.
(a) Before and after each test run, perform a two-point system integrity check using the same procedure as the initial system integrity check described in Section 8.2.5. You may use data from that initial system integrity check as the pre-run data for the first test run, provided it is the most recent system integrity check done before the first run. You may also use the results of a successful post-run system integrity check as the pre-run data for the next test run. Do not make any adjustments to the measurement system during these checks, other than to maintain the target calibration gas flow rate and the proper dilution ratio.
(b) As a time-saving alternative, you may, at the risk of invalidating multiple test runs, skip one or more integrity checks during a test day. Provided there have been no auto-calibrations or other instrument alterations, a single integrity check may suffice as a post-run check to validate (or invalidate) as many consecutive test runs as can be completed during a single test day. All subsequent test days must begin with a pre-run system integrity check subject to the same performance criteria and corrective action requirements as a post-run system integrity check.
(c) Each system integrity check must meet the criteria for system integrity checks in Section 13.2. If a post-run system integrity check is failed, all test runs since the last passed system integrity check are invalid. If a post-run or a pre-run system integrity check is failed, you must take corrective action and pass another 3-point Hg0 system calibration error test (Section 8.2.4) followed by another system integrity check before conducting any additional test runs. Record the results of the pre- and post-run system integrity checks on a form similar to Table 30A-2 in Section 17.0.
8.2.8.2 Drift Check.
Using the data from the successful pre- and post-run system integrity checks, calculate the zero and upscale drift, using Equation 30A-2 in Section 12.3. Exceeding the Section 13.3 specification does not invalidate the run, but corrective action must be taken and a new 3-point Hg0 system calibration error test and a system integrity check must be passed before any more runs are made.
8.3 Dilution-Type Systems—Special Considerations.
When a dilution-type measurement system is used, there are three important considerations that must be taken into account to ensure the quality of the emissions data. First, the critical orifice size and dilution ratio must be selected properly so that the sample dew point will be below the sample line and analyzer temperatures. Second, a high-quality, accurate dilution controller must be used to maintain the correct dilution ratio during sampling. The dilution controller should be capable of monitoring the dilution air pressure, orifice upstream pressure, eductor vacuum, and sample flow rates. Third, differences between the molecular weight of calibration gas mixtures, dilution air, and the stack gas molecular weight must be considered because these can affect the dilution ratio and introduce measurement bias.
8.4 Sampling.
(a) Position the Probe at the first sampling point. Allow the system to flush and equilibrate for at least two times the measurement system response time before recording any data. Then, traverse and record measurements at all required sampling points. Sample at each traverse point for an equal length of time, maintaining the appropriate sample flow rate or dilution ratio (as applicable). For all Hg instrumental method systems, the minimum sampling time at each sampling point must be at least two times the system response time, but not less than 10 minutes. For concentrating systems, the minimum sampling time must also include at least 4 concentration measurement cycles.
(b) After recording data for the appropriate period of time at the first traverse point, you may move the sample Probe to the next point and continue recording, omitting the requirement to allow the system to equilibrate for two times the system response time before recording data at the subsequent traverse points. You must, however, sample at this and all subsequent traverse points for the required minimum amount of time specified in this section. If you must remove the Probe from the stack for any reason, you must again allow the sampling system to equilibrate for at least two times the system response time prior to resuming data recording.
(c) If at any point the measured Hg concentration exceeds the calibration span value, you must at a minimum identify and report this as a deviation from the method. Depending on the data quality objectives of the test, this event may require corrective action before proceeding. If the average Hg concentration for any run exceeds the calibration span value, the run is invalidated.
8.5 Moisture Correction.
If the moisture basis (wet or dry) of the measurements made with this method is different from the moisture basis of either: (1) the applicable emission limit; or (2) a Hg CEMS or sorbent trap monitoring system being evaluated for relative accuracy, you must determine the moisture content of the flue gas and correct the measured gas concentrations to a dry basis using Method 4 in appendix A-3 of this part or other appropriate methods, subject to the approval of the Administrator.
8.6 Optional Interference Test Procedure.
(a) Select an appropriate calibration span that reflects the source(s) to be tested and perform the interference check at 40 percent of the lowest calibration span value anticipated, e.g., 10 μg/m3. Alternatively, successfully conducting the interference test at an absolute Hg concentration of 2μg/m3 will demonstrate performance for an equivalent calibration span of 5 μg/m3, the lowest calibration span allowed for Method 30A testing. Therefore, performing the interference test at the 2 μg/m3 level will serve to demonstrate acceptable performance for all calibration spans greater than or equal to 5 μg/m3.
(b) Introduce the interference test gases listed in Table 30A-3 in Section 17.0 into the measurement system separately or as a mixture. The interference test gases HCl and NO must be introduced as a mixture. The interference test gases must be introduced into the sampling system at the Probe such that the interference gas mixtures pass through all filters, scrubbers, conditioners, and other components as would be configured for normal sampling.
(c) The interference test must be performed using HgCl2, and each interference test gas (or gas mixture) must be evaluated in triplicate. This is accomplished by measuring the Hg response first with only the HgCl2 gas present and then when adding the interference test gas(es) while maintaining the HgCl2 concentration of the test stream constant. It is important that the equipment used to conduct the interference test be of sufficient quality so as to be capable of blending the HgCl2 and interference gases while maintaining the Hg concentration constant. Gas blending system or manifolds may be used.
(d) The duration of each test should be for a sufficient period of time to ensure the Hg measurement system surfaces are conditioned and a stable output is obtained. Measure the Hg response of the analyzer to these gases in μg/m3. Record the responses and determine the overall interference response using Table 30A-4 in Section 17.0 and the equations presented in Section 12.11. The specification in Section 13.4 must be met.
(e) A copy of these data, including the date completed and a signed certification, must be included with each test report. The intent of this test is that the interference test results are intended to be valid for the life of the system. As a result, the Hg measurement system should be operated and tested in a configuration consistent with the configuration that will be used for field applications. However, if the system used for field testing is not consistent with the system that was interference-tested, the interference test must be repeated before it is used for any field applications. Examples of such conditions include, but are not limited to: major changes in dilution ratio (for dilution based systems), changes in catalyst materials, changes in filtering device design or materials, changes in Probe design or configuration, and changes in gas conditioning materials or approaches.
9.0 Quality Control
What quality control measures must I take?
The table which follows is a summary of the mandatory, suggested, and alternative quality assurance and quality control measures and the associated frequency and acceptance criteria. All of the QC data, along with the run data, must be documented and included in the test report.
Summary Table of QA/QC Requirements
| Status1 | Process or Element | QA/QC Specification | Acceptance Criteria | Checking Frequency | |
| S | Identify Data User | Regulatory Agency or other primary end user of data | Before designing test | ||
| M | Analyzer Design | Analyzer range | Sufficiently > than high-level gas to allow determination of system calibration error | ||
| S | Analyzer resolution or sensitivity | < 2.0 % of full-scale range | Manufacturer design | ||
| S | Interference response | Overall response ≤ 3 % of calibration span Alternatively, overall response ≤ 0.3 μg/m3 | |||
| M | calibration Gases | Traceability protocol | Validation of concentration required | ||
| M | High-level Hg0 gas | Equal to the calibration span | Each calibration error test | ||
| M | Mid-level Hg0 gas | 40 to 60% of calibration span | Each calibration error test | ||
| M | Low-level Hg0 gas | 10 to 30% of calibration span | Each calibration error test | ||
| M | High-level HgCl2 gas | Equal to the calibration span | Each system integrity check (if it better represents Cnative than the mid level gas) | ||
| M | Mid-level HgCl2 gas | 40 to 60% of calibration span | Each system integrity check (if it better represents Cnative than the high level gas) | ||
| M | Zero gas | Each system integrity check | |||
| M | Dynamic spike gas (Cnative ≥ 1 μg/m3) | A high-concentration HgCl2 gas, used to produce a spiked sample concentration that is 150 to 200% of the native concentration. | Pre-test; dynamic spiking not required until 1/1/09 | ||
| M | Dynamic spike gas (Cnative < 1 μg/m3) | A high-concentration HgCl2 gas, used to produce a spiked sample concentration that is 1 to 2 μg/m3 above the native concentration. | Pre-test; dynamic spiking not required until 1/1/09 | ||
| S | Data Recorder Design | Data resolution | ≤ 0.5% of full-scale | Manufacturer design | |
| M | Sample Extraction | Probe material | Inert to sample constituents (e.g.,PFA Teflon, or quartz if stack > 500 °F) | Each run | |
| M | Sample Extraction | Probe, filter and sample line temperature | For dry-basis analyzers, keep sample above the dew point, by heating prior to moisture removal For wet-basis analyzers, keep sample above dew point at all times, by heating or dilution | Each run | |
| M | Sample Extraction | calibration valve material | Inert to sample constituents (e.g., PFA Teflon or PFA Teflon coated) | Each test | |
| S | Sample Extraction | sample pump> material | Inert to sample constituents | Each test | |
| M | Sample Extraction | Manifold material | Inert to sample constituents | Each test | |
| M | Particulate Removal | filter inertness | Pass calibration error check | Each calibration error check | |
| M | System calibration Performance | System calibration error (CE) test | CE ≤ 5.0 % of the calibration span for the low-, mid- or high-level Hg0 calibration gas Alternative specification: ≤ 0.5 μg/m3 absolute difference between system response and reference value | Before initial run and after a failed system integrity check or drift test | |
| M | System calibration Performance | System integrity check | Error ≤ 5.0 % of the calibration span for the zero and mid- or high-level HgCl2 calibration gas Alternative specification: ≤ 0.5 μg/m3 absolute difference between system response and reference value | Before initial run, after each run, at the beginning of subsequent test days, and after a failed system integrity check or drift test | |
| M | System Performance | System response time | Used to determine minimum sampling time per point | During initial 3-point system calibration error test | |
| M | System Performance | Drift | ≤ 3.0 % of calibration span for the zero and mid- or high- level gas Alternative specification: ≤ 0.3 μg/m3 absolute difference between pre- and post-run system calibration error percentages | At least once per test day | |
| M | System Performance | Minimum sampling time | The greater of two times the system response time or 10 minutes. Concentrating systems must also include at least 4 cycles | Each sampling point | |
| M | System Performance | Percentage spike recovery and relative standard deviation | Percentage spike recovery, at the target level: 100 ± 10% Relative standard deviation: ≤ 5 percent Alternative specification: absolute difference between calculated and measured spike values ≤ 0.5 μg/m3 | Before initial run; dynamic spiking not required until 1/1/09 | |
| M | Sample Point Selection | Number and Location of Sample Points | For emission testing applications, use 12 points, located according to Method 1 in appendix A-1 to this part, unless the results of a stratification test allow fewer points to be used For Part 60 RATAs, follow the procedures in Performance Specification 2, section 8.1.3, and for Part 75 RATAs, follow the procedures in section 6.5.6 of appendix A to Part 75. That is: • At any test location, you may use 3 sample points located at 16.7, 50.0, and 83.3% of a “long” measurement line passing through the centroidal area; or • At any test location, you may use 6 sample points along a diameter, located according to Method 1 (Part 75 RATAs, only); or • At a location where stratification is not expected and the measurement line is > 2.4 m (7.8 ft), you may use 3 sample points located along a “short” measurement line at 0.4, 1.0, and 2.0 m from the stack or duct wall or, for Part 75 only, sample points may be located at 4.4, 14.6, and 29.6% of the measurement line; or • After a wet scrubber or at a point where dissimilar gas streams are combined, either locate 3 sample points along the “long” measurement line or locate 6 Method 1 points along a diameter (Part 75, only), unless the results of a stratification test allow you to use a “short” 3-point measurement line or to sample at a single point • If it can be demonstrated that stack gas concentration is <3 μg/m3, then the test site is exempted from stratification testing. Use the 3-point “short” measurement line if the stack diameter is > 2.4 m (7.8 ft) and the 3-point “long” line for stack diameters ≤ 2.4 m (7.8 ft) | Prior to first run | |
| A | Sample Point Selection | Stratification Test (see Section 8.1.3) | If the Hg concentration2 at each traverse point during the stratification test is: • Within ± 5% of mean, use 1-point sampling (at the point closest to the mean); or • Not within ± 5% of mean, but is within ± 10% of mean, use 3-point sampling. Locate points according to Section 8.1.3.2.2 of this method Alternatively, if the Hg concentration at each point is: • Within ± 0.2 μg/m3 of mean, use 1-point sampling (at the point closest to the mean); or • Not within ± 0.2 μg/m3 of mean, but is within ± 0.5 μg/m3 of mean, use 3-point sampling. Locate points according to Section 8.1.3.2.2 of this method If the Hg concentration is >10% of the mean at any point, then, if the alternative specification is not met or if the stack diameter is ≤ 2.4 m (7.8 ft): • Perform sampling at 12 Method 1 points; or • Sample at 3 points located at 16.7, 50.0 and 83.3% of the measurement line that exhibited the highest average Hg concentration during stratification test; or • Sample at 6 Method 1 points along the line that exhibited the highest average Hg concentration (Part 75 RATAs, only) | Prior to first run Prior to 1/1/09, you may (1) forgo stratification testing and use 3 sampling points (as per Section 8.1.3.2.2) or (2) perform a SO2 stratification test (see Sections 6.5.6.1 and 6.5.6.3 of appendix A to part 75), in lieu of a Hg stratification test. If the test location is unstratified or minimally stratified for SO2, it can be considered unstratified or minimally stratified for Hg also On and after 1/1/09, only Hg stratification tests are acceptable for the purposes of this method | |
| M | Data Recording | Frequency | Once per cycle | During run | |
| S | Data Parameters | Sample concentration and calibration span | All analyzer readings during each run within calibration span | Each run | |
| M | Data Parameters | Sample concentration and calibration span | All analyzer readings during dynamic spiking tests within 120% of calibration span | Each spike injection | |
| M | Data Parameters | Sample concentration and calibration span | Average Hg concentration for the run ≤ calibration span | Each run |
1 M = Mandatory ; S = Suggested; A = Alternative
2 These may either be the unadjusted Hg concentrations or concentrations normalized to account for temporal variations.
10.0 Calibration and Standardization
What measurement system calibrations are required?
Your analyzer must be calibrated with Hg0 standards. The initial 3-point system calibration error test described in Section 8.2.4 is required before you start the test. Also, prior to and following test runs, the two-point system integrity checks described in Sections 8.2.5 and 8.2.8 are required. On and after January 1, 2009, the pre-test dynamic spiking procedure described in section 8.2.7 is also required to verify that the accuracy of the measurement system is suitable and not adversely affected by the flue gas matrix.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see Section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis listed in this section.
12.1 Nomenclature.
The terms used in the equations are defined as follows:
| Bws | = | Moisture content of sample gas as measured by Method 4 in Appendix A-3 to this part, percent/100. |
| Cavg | = | Average unadjusted Hg concentration for the test run, as indicated by the data recorder μg/m3. |
| Cbaseline | = | Average Hg concentration measured before and after dynamic spiking injections, μg/m3. |
| Cd | = | Hg concentration, dry basis, μg/m3. |
| Cdif | = | Absolute value of the difference between the measured Hg concentrations of the reference HgCl2 calibration gas, with and without the individual or combined interference gases, μg/m3. |
| Cdif avg | = | Average of the 3 absolute values of the difference between the measured Hg concentrations of the reference HgCl2 calibration gas, with and without the individual or combined interference gases, μg/m3. |
| Cgas | = | Average Hg concentration in the effluent gas for the test run, adjusted for system calibration error, μg/m3. |
| Cint | = | Measured Hg concentration of the reference HgCl2 calibration gas plus the individual or combined interference gases, μg/m3. |
| Cm | = | Average of pre- and post-run system integrity check responses for the upscale (i.e., mid- or high-level) calibration gas, μg/m3. |
| Cma | = | Actual concentration of the upscale (i.e., mid- or high-level) calibration gas used for the system integrity checks, μg/m3. |
| C0 | = | Average of pre- and post-run system integrity check responses from the zero gas, μg/m3. |
| Cnative | = | Vapor phase Hg concentration in the source effluent, μg/m3. |
| Cref | = | Measured Hg concentration of the reference HgCl2 calibration gas alone, in the interference test, μg/m3. |
| Cs | = | Measured concentration of a calibration gas (zero-, low-, mid-, or high-level), when introduced in system calibration mode, μg/m3. |
| Cspike | = | Actual Hg concentration of the spike gas, μg/m3. |
| C*spike | = | Hg concentration of the spike gas required to achieve a certain target value for the spiked sample Hg concentration, μg/m3. |
| Css | = | Measured Hg concentration of the spiked sample at the target level, μg/m3. |
| C*ss | = | Expected Hg concentration of the spiked sample at the target level, μg/m3. |
| Ctarget | = | Target Hg concentration of the spiked sample, μg/m3. |
| CTnative | = | Measured tracer gas concentration present in native effluent gas, ppm. |
| CTdir | = | Tracer gas concentration injected with spike gas, ppm. |
| CTv | = | Diluted tracer gas concentration measured in a spiked sample, ppm. |
| Cv | = | Certified Hg0 or HgCl2 concentration of a calibration gas (zero, low, mid, or high), μg/m3. |
| Cw | = | Hg concentration measured under moist sample conditions, wet basis, μg/m3. |
| CS | = | calibration span, μg/m3. |
| D | = | Zero or upscale drift, percent of calibration span. |
| DF | = | Dilution factor of the spike gas, dimensionless. |
| I | = | Interference response, percent of calibration span. |
| QProbe | = | Total flow rate of the stack gas sample plus the spike gas, liters/min. |
| Qspike | = | flow rate of the spike gas, liters/min. |
| Ri | = | Individual injection spike recovery, %. |
| R | = | Mean value of spike recoveries at a particular target level, %. |
| RSD | = | Relative standard deviation, %. |
| SCE | = | System calibration error, percent of calibration span. |
| SCEi | = | Pre-run system calibration error during the two-point system integrity check, percent of calibration span. |
| SCEf | = | Post-run system calibration error during the two-point system integrity check, percent of calibration span. |
12.2 System calibration Error.
Use Equation 30A-1 to calculate the system calibration error. Equation 30A-1 applies to: 3-point system calibration error tests performed with Hg0 standards; and pre- and post-run two-point system integrity checks performed with HgCl2.
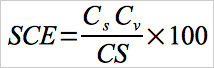 Eq.30A-1
Eq.30A-1
12.3 Drift Assessment.
Use Equation 30A-2 to separately calculate the zero and upscale drift for each test run.
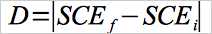 Eq.30A-2
Eq.30A-2
12.3 Effluent Hg Concentration.
For each test run, calculate Cavg, the arithmetic average of all valid Hg concentration values recorded during the run. Then, adjust the value of Cavg for system calibration error, using Equation 30A-3.
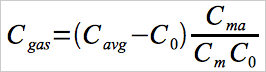 Eq. 30A-3
Eq. 30A-3
12.4 Moisture Correction.
Use Equation 30A-4a if your measurements need to be corrected to a dry basis.
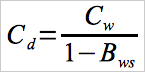 Eq.30A-4a
Eq.30A-4a
Use Equation 30A-4b if your measurements need to be corrected to a wet basis.
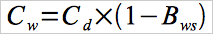 Eq.30A-4b
Eq.30A-4b
12.5 Dynamic Spike Gas Concentrations.
Use Equation 30A-5 to determine the spike gas concentration needed to produce a spiked sample with a certain “target” Hg concentration.
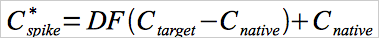 Eq. 30A-5
Eq. 30A-5
12.6 Spiked Sample Concentration.
Use Equation 30A-6 to determine the expected or theoretical Hg concentration of a spiked sample.
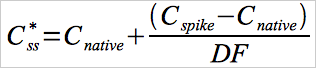 Eq.30A-6
Eq.30A-6
12.7 Spike Recovery.
Use Equation 30A-7 to calculate the percentage recovery of each spike.
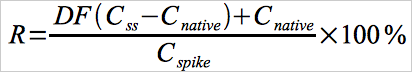 Eq.30A-7
Eq.30A-7
12.8 Relative Standard Deviation.
Use Equation 30A-8 to calculate the relative standard deviation of the individual percentage spike recovery values from the mean.
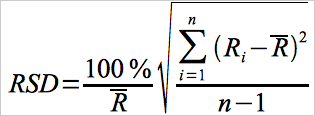 Eq. 30A-8
Eq. 30A-8
12.9 Spike Dilution Factor.
Use Equation 30A-9 to calculate the spike dilution factor, using either direct flow measurements or tracer gas measurements.
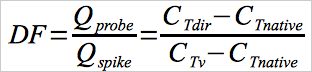 Eq. 30A-9
Eq. 30A-9
12.10 Native Concentration.
For spiking procedures that inject blank or carrier gases (at the spiking flow rate, Qspike) between spikes, use Equation 30A-10 to calculate the native concentration.
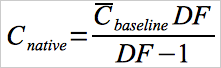 Eq. 30A-10
Eq. 30A-10
For spiking procedures that halt all injections between spikes, the native concentration equals the average baseline concentration (see Equation 30A-11).
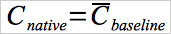 Eq. 30A-11
Eq. 30A-11
12.11 Overall Interference Response.
Use equation 30A-12 to calculate the overall interference response.
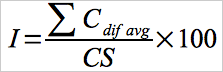 Eq. 30A-12
Eq. 30A-12
Where, for each interference gas (or mixture):
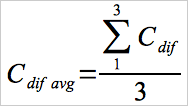 Eq.30A-13
Eq.30A-13
and
 Eq. 30A-14
Eq. 30A-14
13.0 Method Performance
13.1 System calibration Error Test.
This specification applies to the 3-point system calibration error tests using Hg0. At each calibration gas level tested (low-, mid-, or high-level), the calibration error must be within ± 5.0 percent of the calibration span. Alternatively, the results are acceptable if | Cs - Cv | ≤ 0.5 μg/m3.
13.2 System Integrity Checks.
This specification applies to all pre- and post-run 2-point system integrity checks using HgCl2 and zero gas. At each calibration gas level tested (zero and mid- or high-level), the error must be within ± 5.0 percent of the calibration span. Alternatively, the results are acceptable if | Cs - Cv | ≤ 0.5 μg/m3.
13.3 Drift.
For each run, the low-level and upscale drift must be less than or equal to 3.0 percent of the calibration span. The drift is also acceptable if the pre- and post-run system integrity check responses do not differ by more than 0.3 μg/m3 (i.e., | Cs post-run - Cs pre-run | ≤ 0.3 μg/m3).
13.4 Interference Test.
Summarize the results following the format contained in Table 30A-4. For each interference gas (or mixture), calculate the mean difference between the measurement system responses with and without the interference test gas(es). The overall interference response for the analyzer that was used for the test (calculated according to Equation 30A-12), must not be greater than 3.0 percent of the calibration span used for the test (see Section 8.6). The results of the interference test are also acceptable if the sum of the absolute average differences for all interference gases (i.e., Σ Cdif avg) does not exceed 0.3 μg/m3.
13.5 Dynamic Spiking Test.
For the pre-test dynamic spiking, the mean value of the percentage spike recovery must be 100 ± 10 percent. In addition, the relative standard deviation (RSD) of the individual percentage spike recovery values from the mean must be ≤ 5.0 percent. Alternatively, if the mean percentage recovery is not met, the results are acceptable if the absolute difference between the theoretical spiked sample concentration (see Section 12.6) and the actual average value of the spiked sample concentration is ≤ 0.5 μg/m3.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. EPA Traceability Protocol for Qualification and Certification of Elemental Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
2. EPA Traceability Protocol for Qualification and Certification of Oxidized Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
3. EPA Traceability Protocol for Assay and Certification of Gaseous calibration Standards, expected revision publication date December 2008, see www.epa.gov/ttn/emc.
17.0 Figures and Tables
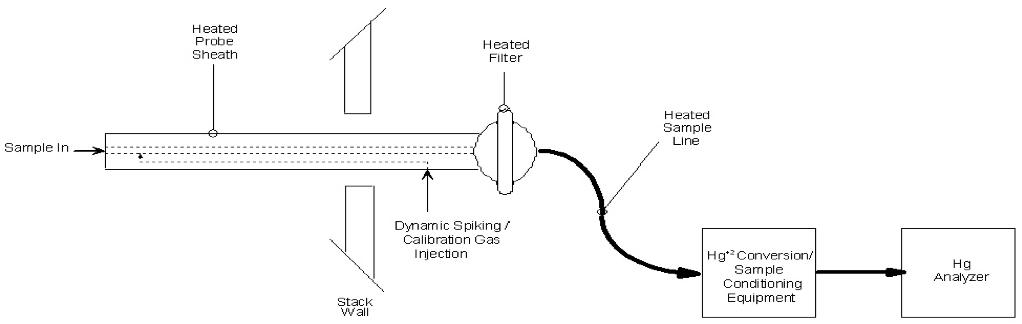
Figure 30A-1: Example Hg Measurement System
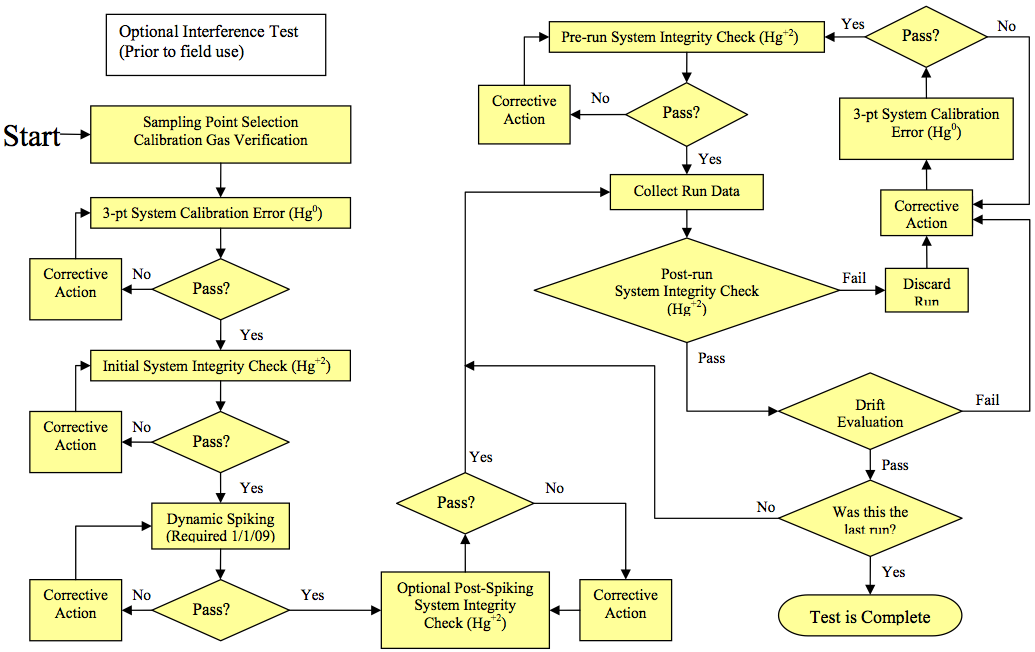
Figure 30A-2. Testing flow Chart
Table 30A-1: 3-Point System calibration Error Tests
Tables and Prinouts
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources