- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 25d - Determination Of The Volatile Organic Concentration Of Waste Samples
NOTE: Performance of this method should not be attempted by persons unfamiliar with the operation of a flame ionization detector (FID) or an electrolytic conductivity detector (ELCD) because knowledge beyond the scope of this presentation is required.
1.0 Scope and Application.
1.1 Analyte. Volatile Organic Compounds. No CAS No. assigned.
1.2 Applicability. This method is applicable for determining the volatile organic (VO) concentration of a waste sample.
2.0 Summary of Method.
2.1 Principle. A sample of waste is obtained at a point which is most representative of the unexposed waste (where the waste has had minimum opportunity to volatilize to the atmosphere). The sample is suspended in an organic/aqueous matrix, then heated and purged with nitrogen for 30 min in order to separate certain organic compounds. Part of the sample is analyzed for carbon concentration, as methane, with an FID, and part of the sample is analyzed for chlorine concentration, as chloride, with an ELCD. The VO concentration is the sum of the carbon and chlorine content of the sample.
3.0 Definitions.
3.1 Well-mixed in the context of this method refers to turbulent flow which results in multiple-phase waste in effect behaving as single-phase waste due to good mixing.
4.0 Interferences. [Reserved]
5.0 Safety.
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
6.0 Equipment and Supplies.
NOTE: Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
6.1 Sampling. The following equipment is required:
6.1.1 Sampling Tube. Flexible Teflon, 0.25 in. ID (6.35 mm).
6.1.2 Sample Container. Borosilicate glass, 40-mL, and a Teflon-lined screw cap capable of forming an air tight seal.
6.1.3 Cooling Coil. Fabricated from 0.25 in (6.35 mm). ID 304 stainless steel tubing with a thermocouple at the coil outlet.
6.2 Analysis.
The following equipment is required.
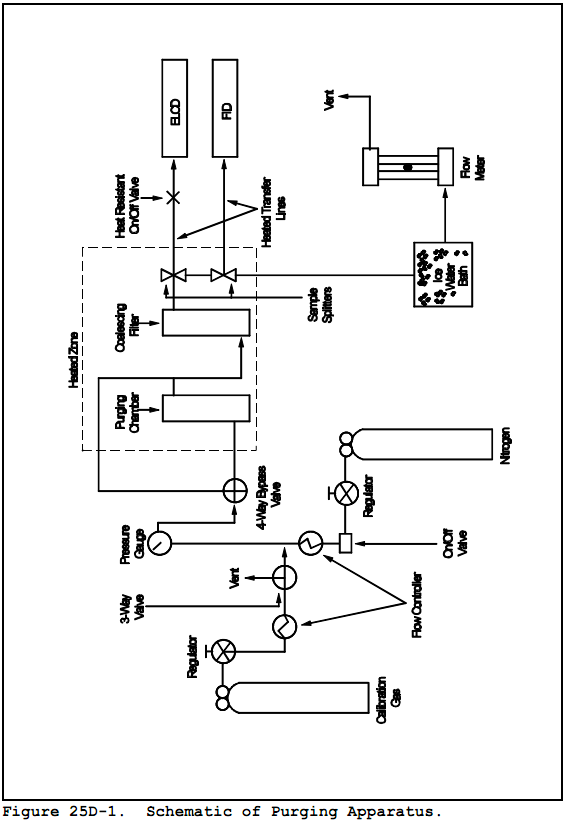
6.2.1 Purging Apparatus.
For separating the VO from the waste sample. A schematic of the system is shown in Figure 25D-1. The purging apparatus consists of the following major components.
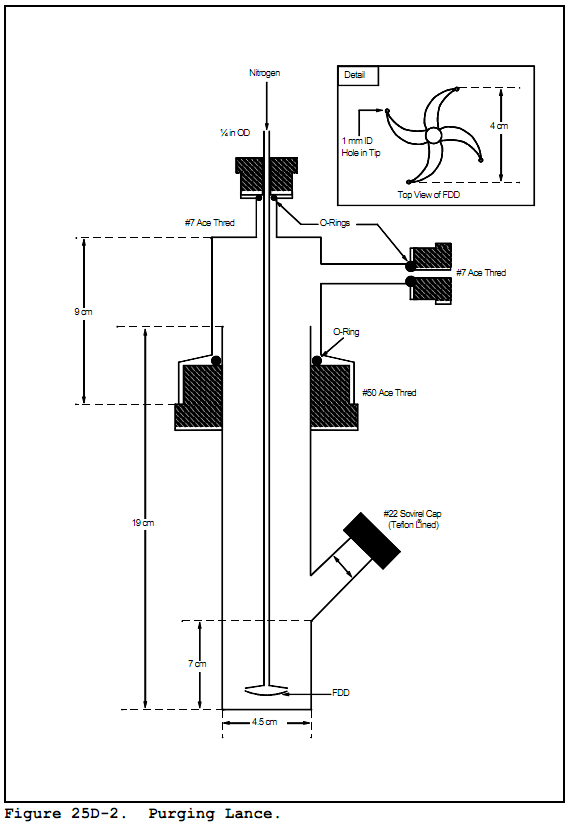
6.2.1.1 Purging Flask. A glass container to hold the sample while it is heated and purged with dry nitrogen. The cap of the purging flask is equipped with three fittings: one for a purging lance (fitting with the #7 Ace-thread), one for the Teflon exit tubing (side fitting, also a #7 Ace-thread), and a third (a 50-mm Ace-thread) to attach the base of the purging flask as shown in Figure 25D-2. The base of the purging flask is a 50-mm ID (2 in) cylindrical glass tube. One end of the tube is open while the other end is sealed. Exact dimensions are shown in Figure 25D-2.
6.2.1.2 Purging Lance. glass tube, 6-mm OD (0.2 in) by 30 cm (12 in) long. The purging end of the tube is fitted with a four-arm bubbler with each tip drawn to an opening 1 mm (0.04 in) in diameter. Details and exact dimensions are shown in Figure 25D-2.
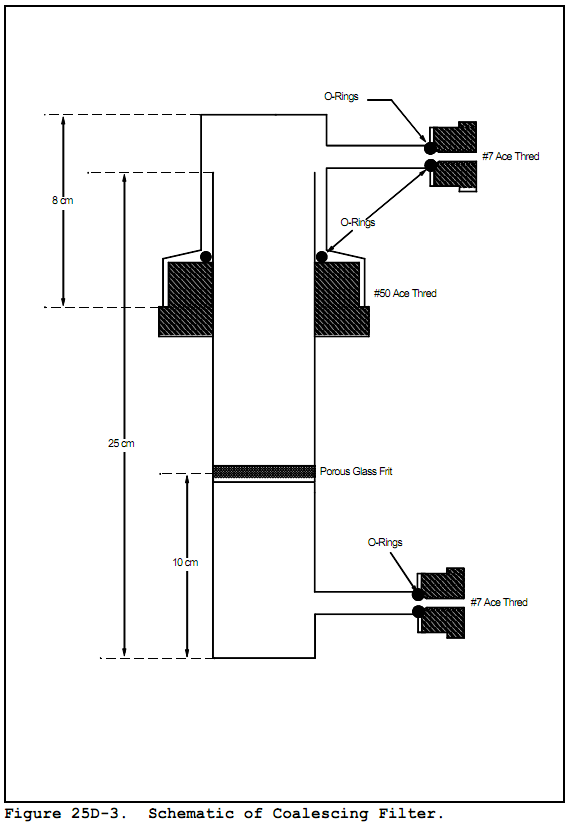
6.2.1.3 Coalescing filter. Porous fritted disc incorporated into a container with the same dimensions as the purging flask. The details of the design are shown in Figure 25D-3.
6.2.1.4 Constant temperature Chamber. A forced draft oven capable of maintaining a uniform temperature around the purging flask and coalescing filter of 75 + 2°C (167 ± 3.6°F).
6.2.1.5 Three-way Valve. Manually operated, stainless steel. To introduce calibration gas into system.
6.2.1.6 flow Controllers. Two, adjustable. One capable of maintaining a purge gas flow rate of 6 + 0.06 L/min (0.2 ± 0.002 ft3/min) The other capable of maintaining a calibration gas flow rate of 1-100 mL/min (0.00004-0.004 ft3/min).
6.2.1.7 Rotameter. For monitoring the air flow through the purging system (0-10 L/min)(0-0.4 ft3/min).
6.2.1.8 Sample Splitters. Two heated flow restrictors (placed inside oven or heated to 120 + 10°C (248 + 18 °F) ). At a purge rate of 6 L/min (0.2 ft3/min), one will supply a constant flow to the first detector (the rest of the flow will be directed to the second sample splitter). The second splitter will split the analytical flow between the second detector and the flow restrictor. The approximate flow to the FID will be 40 mL/min (0.0014 ft3/min) and to the ELCD will be 15 mL/min (0.0005 ft3/min), but the exact flow must be adjusted to be compatible with the individual detector and to meet its linearity requirement. The two sample splitters will be connected to each other by 1/8" OD (3.175 mm) stainless steel tubing.
6.2.1.9 flow Restrictor. Stainless steel tubing, 1/8" OD (3.175 mm), connecting the second sample splitter to the ice bath. Length is determined by the resulting pressure in the purging flask (as measured by the pressure gauge). The resulting pressure from the use of the flow restrictor shall be 6-7 psig.
6.2.1.10 filter Flask. With one-hole stopper. Used to hold ice bath. Excess purge gas is vented through the flask to prevent condensation in the flowmeter and to trap volatile organic compounds.
6.2.1.11 Four-way Valve. Manually operated, stainless steel. Placed inside oven, used to bypass purging flask.
6.2.1.12 On/Off Valves. Two, stainless steel. One heat resistant up to 130°C (266 °F) and placed between oven and ELCD. The other a toggle valve used to control purge gas flow.
6.2.1.13 Pressure Gauge. Range 0-40 psi. To monitor pressure in purging flask and coalescing filter.
6.2.1.14 Sample Lines. Teflon, 1/4" OD (6.35 mm), used inside the oven to carry purge gas to and from purging chamber and to and from coalescing filter to four-way valve. Also used to carry sample from four-way valve to first sample splitter.
6.2.1.15 Detector tubing. Stainless steel, 1/8" OD (3.175 mm), heated to 120 + 10°C (248 ± 18 °F) . Used to carry sample gas from each sample splitter to a detector. Each piece of tubing must be wrapped with heat tape and insulating tape in order to insure that no cold spots exist. The tubing leading to the ELCD will also contain a heat- resistant on-off valve (Section 6.2.1.12) which shall also be wrapped with heat-tape and insulation.
6.2.2 Volatile Organic Measurement System.
Consisting of an FID to measure the carbon concentration of the sample and an ELCD to measure the chlorine concentration.
6.2.2.1 FID. A heated FID meeting the following specifications is required.
6.2.2.1.1 Linearity. A linear response (+ 5 percent)
over the operating range as demonstrated by the procedures established in Section 10.1.1.
6.2.2.1.2 Range. A full scale range of 50 pg carbon/sec to 50 μg carbon/sec. Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.2.2.1.3 Data Recording System. A digital integration system compatible with the FID for permanently recording the output of the detector. The recorder shall have the capability to start and stop integration at points selected by the operator or it shall be capable of the "integration by slices" technique (this technique involves breaking down the chromatogram into smaller increments, integrating the area under the curve for each portion, subtracting the background for each portion, and then adding all of the areas together for the final area count).
6.2.2.2 ELCD. An ELCD meeting the following specifications is required. 1-propanol must be used as the electrolyte. The electrolyte flow through the conductivity cell shall be 1 to 2 mL/min (0.00004 to 0.00007 ft3/min).
NOTE: A 1/4-in. ID (6.35 mm) quartz reactor tube is strongly recommended to reduce carbon buildup and the resulting detector maintenance.
6.2.2.2.1 Linearity. A linear response (+ 10 percent) over the response range as demonstrated by the procedures in Section 10.1.2.
6.2.2.2.2 Range. A full scale range of 5.0 pg/sec to 500 ng/sec chloride. Signal attenuators shall be available to produce a minimum signal response of 10 percent of full scale.
6.2.2.2.3 Data Recording System. A digital integration system compatible with the output voltage range of the ELCD. The recorder must have the capability to start and stop integration at points selected by the operator or it shall be capable of performing the "integration by slices" technique.
7.0 Reagents and Standards.
7.1 Sampling.
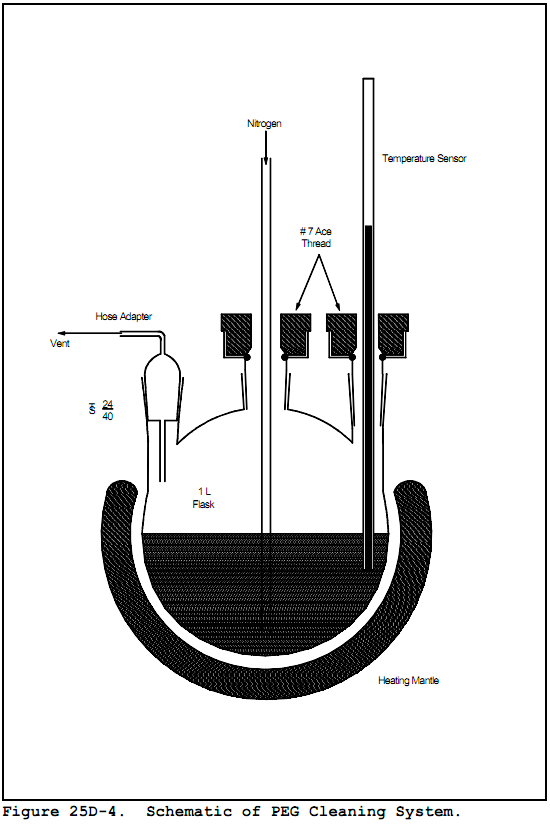
7.1.1 Polyethylene Glycol (PEG). Ninety-eight percent pure with an average molecular weight of 400. Before using the PEG, remove any organic compounds that might be detected as volatile organics by heating it to 120°C (248 °F) and purging it with nitrogen at a flow rate of 1 to 2 L/min (0.04 to 0.07 ft3/min) for 2 hours. The cleaned PEG must be stored under a 1 to 2 L/min (0.04 to 0.07 ft3/min) nitrogen purge until use. The purge apparatus is shown in Figure 25D-4.
7.2 Analysis.
7.2.1 Sample Separation. The following are required for the sample purging step.
7.2.1.1 PEG. Same as Section 7.1.1.
7.2.1.2 Purge Gas. Zero grade nitrogen (N2), containing less than 1 ppm carbon.
7.2.2 Volatile Organics Measurement. The following are required for measuring the VO concentration.
7.2.2.1 Hydrogen (H2). Zero grade H2, 99.999 percent pure.
7.2.2.2 Combustion Gas. Zero grade air or oxygen as required by the FID.
7.2.2.3 calibration Gas. Pressurized gas cylinder containing 10 percent propane and 1 percent 1,1-dichloroethylene by volume in nitrogen.
7.2.2.4 Water. Deionized distilled water that conforms to American Society for Testing and Materials Specification D 1193-74, Type 3, is required for analysis. At the option of the analyst, the KMnO4 test for oxidizable organic matter may be omitted when high concentrations are not expected to be present.
7.2.2.5 1-Propanol. ACS grade or better. Electrolyte Solution. For use in the ELCD.
7.3 Quality Assurance Audit Samples.
7.3.1 It is recommended, but not required, that a
performance audit sample be analyzed in conjunction with the field samples. The audit sample should be in a suitable sample matrix at a concentration similar to the actual field samples.
7.3.2 When making compliance determinations, and upon availability, audit samples may be obtained from the appropriate EPA regional Office or from the responsible enforcement authority and analyzed in conjunction with the field samples.
NOTE: The responsible enforcement authority should be notified at least 30 days prior to the test date to allow sufficient time for sample delivery.
8.0 Sample Collection, Preservation, Storage, and Transport.
8.1 Sampling.
8.1.1 Sampling Plan Design and Development. Use the procedures in chapter nine of Reference 1 in Section 16 as guidance in developing a sampling plan.
8.1.2 Single Phase or Well-mixed Waste.
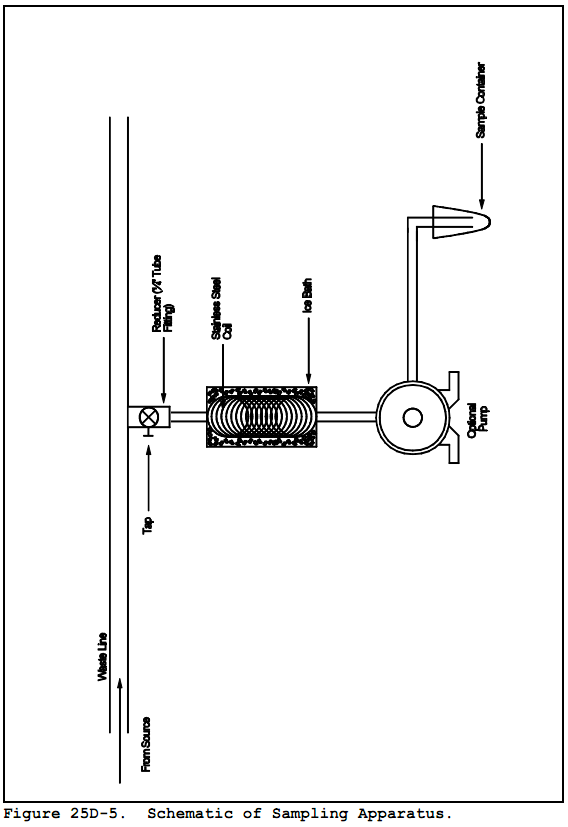
8.1.2.1 Install a sampling tap to obtain the sample at a point which is most representative of the unexposed waste (where the waste has had minimum opportunity to volatilize to the atmosphere). Assemble the sampling apparatus as shown in Figure 25D-5.
8.1.2.2 Prepare the sampling containers as follows: Pour 30 mL of clean PEG into the container. PEG will reduce but not eliminate the loss of organics during sample collection. Weigh the sample container with the screw cap, the PEG, and any labels to the nearest 0.01 g and record the weight (mst). Store the containers in an ice bath until 1 hour before sampling (PEG will solidify at ice bath temperatures; allow the containers to reach room temperature before sampling).
8.1.2.3 Begin sampling by purging the sample lines and cooling coil with at least four volumes of waste. Collect the purged material in a separate container and dispose of it properly.
8.1.2.4 After purging, stop the sample flow and direct the sampling tube to a preweighed sample container, prepared as described in Section 8.1.2.2. Keep the tip of the tube below the surface of the PEG during sampling to minimize contact with the atmosphere. Sample at a flow rate such that the temperature of the waste is less than 10°C (50 °F). Fill the sample container and immediately cap it (within 5 seconds) so that a minimum headspace exists in the container. Store immediately in a cooler and cover with ice.
8.1.3 Multiple-phase Waste. Collect a 10 g sample of each phase of waste generated using the procedures described in Section 8.1.2 or 8.1.5. Each phase of the waste shall be analyzed as a separate sample. Calculate the weighted average VO concentration of the waste using Equation 25D-13 (Section 12.14).
8.1.4 Solid waste. Add approximately 10 g of the solid waste to a container prepared in the manner described in Section 8.1.2.2, minimizing headspace. Cap and chill immediately.
8.1.5 Alternative to Tap Installation. If tap installation is impractical or impossible, fill a large, clean, empty container by submerging the container into the waste below the surface of the waste. Immediately fill a container prepared in the manner described in Section 8.1.2.2 with approximately 10 g of the waste collected in the large container. Minimize headspace, cap and chill immediately.
8.1.6 Alternative sampling techniques may be used upon the approval of the Administrator.
8.2 Sample Recovery.
8.2.1 Assemble the purging apparatus as shown in Figures 25D-1 and 25D-2. The oven shall be heated to 75 + 2°C (167 ± 3.6 °F). The sampling lines leading from the oven to the detectors shall be heated to 120 + 10°C (248 ± 18 °F) with no cold spots. The flame ionization detector shall be operated with a heated block. Adjust the purging lance so that it reaches the bottom of the chamber.
8.2.2 Remove the sample container from the cooler, and wipe the exterior of the container to remove any extraneous ice, water, or other debris. Reweigh the sample container to the nearest 0.01 g, and record the weight (msf). Pour the contents of the sample container into the purging flask, rinse the sample container three times with a total of 20 mL of PEG (since the sample container originally held 30 mL of PEG, the total volume of PEG added to the purging flask will be 50 mL), transferring the rinsings to the purging flask after each rinse. Cap purging flask between rinses. The total volume of PEG in the purging flask shall be 50 mL. Add 50 mL of water to the purging flask.
9.0 Quality Control.
9.1 Quality Control Samples.
If audit samples are not available, prepare and analyze the two types of quality control samples (QCS) listed in Sections 9.4.1 and 9.4.2. Before placing the system in operation, after a shutdown of greater than six months, and after any major modifications, analyze each QCS in triplicate. For each detector, calculate the percent recovery by dividing measured concentration by theoretical concentration and multiplying by 100. Determine the mean percent recovery for each detector for each QCS triplicate analysis. The RSD for any triplicate analysis shall be <10 percent. For QCS 1 (methylene chloride), the percent recovery shall be >90 percent for carbon as methane, and >55 percent for chlorine as chloride. For QCS 2 (1,3-dichloro-2-propanol), the percent recovery shall be <15 percent for carbon as methane, and <6 percent for chlorine as chloride. If the analytical system does not meet the above-mentioned criteria for both detectors, check the system parameters (temperature, system pressure, purge rate, etc.), correct the problem, and repeat the triplicate analysis of each QCS.
9.1.1 QCS 1, Methylene Chloride. Prepare a stock solution by weighing, to the nearest 0.1 mg, 55 μL of HPLC grade methylene chloride in a tared 5 mL volumetric flask. Record the weight in milligrams, dilute to 5 mL with cleaned PEG, and inject 100 μL of the stock solution into a sample prepared as a water blank (50 mL of cleaned PEG and 60 mL of water in the purging flask). Analyze the QCS according to the procedures described in Sections 10.2 and 10.3, excluding Section 10.2.2. To calculate the theoretical carbon concentration (in mg) in QCS 1, multiply mg of methylene chloride in the stock solution by 3.777 x 10-3. To calculate the theoretical chlorine concentration (in mg) in QCS 1, multiply mg of methylene chloride in the stock solution by 1.670 x 10-2.
9.1.2 QCS 2, 1,3-dichloro-2-propanol. Prepare a stock solution by weighing, to the nearest 0.1 mg, 60 μL of high purity grade 1,3-dichloro-2-propanol in a tared 5 mL volumetric flask. Record the weight in milligrams, dilute to 5 mL with cleaned PEG, and inject 100 μL of the stock solution into a sample prepared as a water blank (50 mL of cleaned PEG and 60 mL of water in the purging flask). Analyze the QCS according to the procedures described in Sections 10.2 and 10.3, excluding Section 10.2.2. To calculate the theoretical carbon concentration (in mg) in QCS 2, multiply mg of 1,3-dichloro-2-propanol in the stock solution by 7.461 x 10-3. To calculate the theoretical chlorine concentration (in mg) in QCS 2, multiply mg of 1,3-dichloro-2-propanol in the stock solution by 1.099 x 10-2.
9.1.3 Routine QCS Analysis. For each set of compliance samples (in this context, set is per facility, per compliance test), analyze one QCS 1 and one QCS 2 sample. The percent recovery for each sample for each detector shall be + 13 percent of the mean recovery established for the most recent set of QCS triplicate analysis (Section 9.4). If the sample does not meet this criteria, check the system components and analyze another QCS 1 and 2 until a single set of QCS meet the + 13 percent criteria.
10.0 Calibration and Standardization.
10.1 Initial Performance Check of Purging System.
Before placing the system in operation, after a shutdown of greater than six months, after any major modifications, and at least once per month during continuous operation, conduct the linearity checks described in Sections 10.1.1 and 10.1.2. Install calibration gas at the three-way calibration gas valve. See Figure 25D-1.
10.1.1 Linearity Check Procedure.
Using the calibration standard described in Section 7.2.2.3 and by varying the injection time, it is possible to calibrate at multiple concentration levels. Use Equation 25D-3 to calculate three sets of calibration gas flow rates and run times needed to introduce a total mass of carbon, as methane, (mc) of 1, 5, and 10 mg into the system (low, medium and high FID calibration, respectively). Use Equation 25D-4 to calculate three sets of calibration gas flow rates and run times needed to introduce a total chloride mass (mch) of 1, 5, and 10 mg into the system (low, medium and high ELCD calibration, respectively). With the system operating in standby mode, allow the FID and the ELCD to establish a stable baseline. Set the secondary pressure regulator of the calibration gas cylinder to the same pressure as the purge gas cylinder and set the proper flow rate with the calibration flow controller (see Figure 25D- 1). The calibration gas flow rate can be measured with a flowmeter attached to the vent position of the calibration gas valve. Set the four-way bypass valve to standby position so that the calibration gas flows through the coalescing filter only. Inject the calibration gas by turning the calibration gas valve from vent position to inject position. Continue the calibration gas flow for the appropriate period of time before switching the calibration valve to vent position. Continue recording the response of the FID and the ELCD for 5 min after switching off calibration gas flow. Make triplicate injections of all six levels of calibration.
10.1.2 Linearity Criteria.
Calculate the average response factor (Equations 25D-5 and 25D-6) and the relative standard deviation (RSD) (Equation 25D-10) at each level of the calibration curve for both detectors. Calculate the overall mean of the three response factor averages for each detector. The FID linearity is acceptable if each response factor is within 5 percent of the overall mean and if the RSD for each set of triplicate injections is less than 5 percent. The ELCD linearity is acceptable if each response factor is within 10 percent of the overall mean and if the RSD for each set of triplicate injections is less than 10 percent. Record the overall mean value of the response factors for the FID and the ELCD. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat Sections 10.1.1 and 10.1.2.
10.2 Daily calibrations.
10.2.1 Daily Linearity Check.
Follow the procedures outlined in Section 10.1.1 to analyze the medium level calibration for both the FID and the ELCD in duplicate at the start of the day. Calculate the response factors and the RSDs for each detector. For the FID, the calibration is acceptable if the average response factor is within 5 percent of the overall mean response factor (Section 10.1.2) and if the RSD for the duplicate injection is less than 5 percent. For the ELCD, the calibration is acceptable if the average response factor is within 10 percent of the overall mean response factor (Section 10.1.2) and if the RSD for the duplicate injection is less than 10 percent. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat Sections 10.1.1 and 10.1.2.
10.2.2 Calibration Range Check.
10.2.2.1 If the waste concentration for either detector falls below the range of calibration for that detector, use the procedure outlined in Section 10.1.1 to choose two calibration points that bracket the new target concentration. Analyze each of these points in triplicate (as outlined in Section 10.1.1) and use the criteria in Section 10.1.2 to determine the linearity of the detector in this "mini-calibration" range.
10.2.2.2 After the initial linearity check of the mini-calibration curve, it is only necessary to test one of the points in duplicate for the daily calibration check (in addition to the points specified in Section 10.2.1). The average daily mini-calibration point should fit the linearity criteria specified in Section 10.2.1. If the calibration for either the FID or the ELCD does not meet the criteria, correct the detector/system problem and repeat the calibration procedure mentioned in the first paragraph of Section 10.2.2. A mini-calibration curve for waste concentrations above the calibration curve for either detector is optional.
10.3 Analytical Balance.
Calibrate against standard weights.
11.0 Analysis.
11.1 Sample Analysis.
11.1.1 Turn on the constant temperature chamber and allow the temperature to equilibrate at 75 + 2°C (167 ± 3.6 °F). Turn the four-way valve so that the purge gas bypasses the purging flask, the purge gas flowing through the coalescing filter and to the detectors (standby mode). Turn on the purge gas. Allow both the FID and the ELCD to warm up until a stable baseline is achieved on each detector. Pack the filter flask with ice. Replace ice after each run and dispose of the waste water properly. When the temperature of the oven reaches 75 + 2°C (167 ± 3.6 °F), start both integrators and record baseline. After 1 min, turn the four-way valve so that the purge gas flows through the purging flask, to the coalescing filter and to the sample splitters (purge mode). Continue recording the response of the FID and the ELCD. Monitor the readings of the pressure gauge and the rotameter. If the readings fall below established setpoints, stop the purging, determine the source of the leak, and resolve the problem before resuming. Leaks detected during a sampling period invalidate that sample.
11.1.2 As the purging continues, monitor the output of the detectors to make certain that the analysis is proceeding correctly and that the results are being properly recorded. Every 10 minutes read and record the purge flow rate, the pressure and the chamber temperature. Continue the purging for 30 minutes.
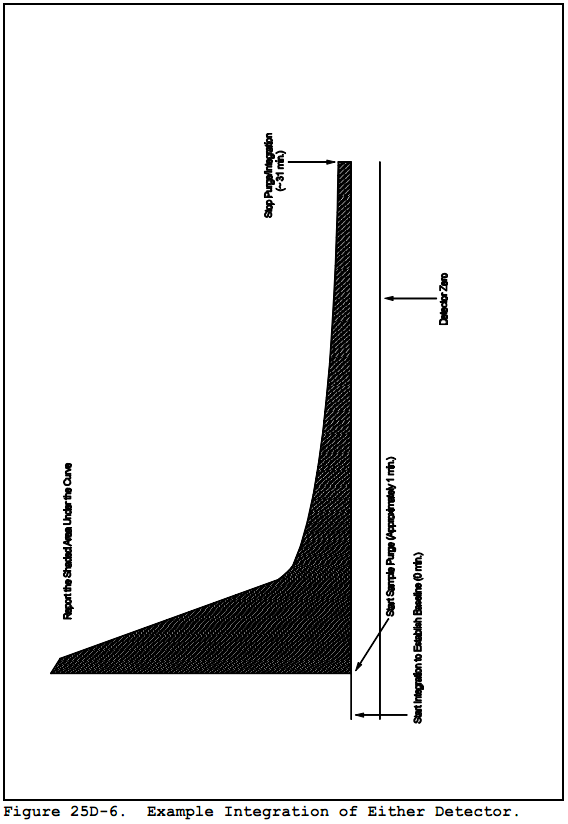
11.1.3 For each detector output, integrate over the entire area of the peak starting at 1 minute and continuing until the end of the run. Subtract the established baseline area from the peak area. Record the corrected area of the peak. See Figure 25D-6 for an example integration.
11.2 Water Blank.
A water blank shall be analyzed for each batch of cleaned PEG prepared. Transfer about 60 mL of water into the purging flask. Add 50 mL of the cleaned PEG to the purging flask. Treat the blank as described in Sections 8.2 and 8.3, excluding Section 8.2.2. Calculate the concentration of carbon and chlorine in the blank sample (assume 10 g of waste as the mass). A VO concentration equivalent to <10 percent of the applicable standard may be subtracted from the measured VO concentration of the waste samples. Include all blank results and documentation in the test report.
11.3 Audit Sample Analysis.
11.3.1 When the method is used to analyze samples to demonstrate compliance with a source emission regulation, an audit sample, if available, must be analyzed.
11.3.2 Concurrently analyze the audit sample and the compliance samples in the same manner to evaluate the technique of the analyst and the standards preparation.
11.3.3 The same analyst, analytical reagents, and analytical system must be used for the compliance samples and the audit sample. If this condition is met, duplicate auditing of subsequent compliance analyses for the same enforcement agency within a 30-day period is waived. An audit sample may not be used to validate different sets of compliance samples under the jurisdiction of separate enforcement agencies, unless prior arrangements have been made with both enforcement agencies.
11.4 Audit Sample Results.
11.4.1 Calculate the audit sample concentrations and submit results using the instructions provided with the audit samples.
11.4.2 Report the results of the audit samples and the compliance determination samples along with their identification numbers, and the analyst's name to the responsible enforcement authority. Include this information with reports of any subsequent compliance analyses for the same enforcement authority during the 30-day period.
12.0 Data Analysis and Calculations.
12.1 Nomenclature.
| Ab | = | Area under the water blank response curve, counts. |
| Ac | = | Area under the calibration response curve, counts. |
| As | = | Area under the sample response curve, counts. |
| C | = | Concentration of volatile organics in the sample, ppmw. |
| Cc | = | Concentration of carbon, as methane, in the calibration gas, mg/L. |
| Cch | = | Concentration of chloride in the calibration gas, mg/L. |
| Cj | = | VO concentration of phase j, ppmw. |
| DRt | = | Average daily response factor of the FID, mg CH4/counts. |
| Drth | = | Average daily response factor of the ELCD, mg Cl-/counts. |
| Fj | = | Weight fraction of phase j present in the waste. |
| mc | = | Mass of carbon, as methane, in a calibration run, mg. |
| mch | = | Mass of chloride in a calibration run, mg. |
| ms | = | Mass of the waste sample, g. |
| msc | = | Mass of carbon, as methane, in the sample, mg. |
| msf | = | Mass of sample container and waste sample, g. |
| msh | = | Mass of chloride in the sample, mg. |
| mst | = | Mass of sample container prior to sampling, g. |
| mVO | = | Mass of volatile organics in the sample, mg. |
| n | = | Total number of phases present in the waste. |
| Pp | = | Percent propane in calibration gas (L/L). |
| Pvc | = | Percent 1,1-dichloroethylene in calibration gas (L/L). |
| Qc | = | Flow rate of calibration gas, L/min. |
| tc | = | Length of time standard gas is delivered to the analyzer, min. |
| W | = | Weighted average VO concentration, ppmw. |
12.2 Concentration of Carbon, as Methane, in the calibration gas.
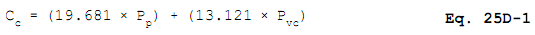
12.3 Concentration of Chloride in the calibration gas.

12.4 Mass of Carbon, as Methane, in a calibration run.

12.5 Mass of Chloride in a calibration run.

12.6 FID Response Factor, mg/counts.

12.7 ELCD Response Factor, mg/counts.

12.8 Mass of Carbon in the Sample.

12.9 Mass of Chloride in the Sample.

12.10 Mass of Volatile Organics in the Sample.

12.11 Relative Standard Deviation.
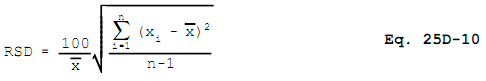
12.12 Mass of Sample.

12.13 Concentration of Volatile Organics in Waste.

12.14 Weighted Average VO Concentration of Multi- phase Waste.

13.0 Method Performance. [Reserved]
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
- "Test Methods for Evaluating Solid Waste, Physical/Chemistry Methods", U.S. Environmental Protection Agency. Publication SW-846, 3rd Edition, November 1986 as amended by Update I, November 1990.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources