- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 18 - Measurement Of Gaseous Organic Compound Emissions By Gas Chromatography
NOTE: This method is not inclusive with respect to specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3.
NOTE: This method should not be attempted by persons unfamiliar with the performance characteristics of gas chromatography, nor by those persons who are unfamiliar with source sampling. Particular care should be exercised in the area of safety concerning choice of equipment and operation in potentially explosive atmospheres.
Content [ show/hide ].
1.0 Scope and Application.
1.1 Analyte.
Total gaseous organic compounds.
1.2 Applicability.
1.2.1 This method is designed to measure gaseous organics emitted from an industrial source. While designed for ppm level sources, some detectors are quite capable of detecting compounds at ambient levels, e.g., ECD, ELCD, and helium ionization detectors. Some other types of detectors are evolving such that the sensitivity and applicability may well be in the ppb range in only a few years.
1.2.2 This method will not determine compounds that (1) are polymeric (high molecular weight), (2) can polymerize before analysis, or (3) have very low vapor pressures at stack or instrument conditions.
1.3 Range.
The lower range of this method is determined by the sampling system; adsorbents may be used to concentrate the sample, thus lowering the limit of detection below the 1 part per million (ppm) typically achievable with direct interface or bag sampling. The upper limit is governed by GC detector saturation or column overloading; the upper range can be extended by dilution of sample with an inert gas or by using smaller volume gas sampling loops. The upper limit can also be governed by condensation of higher boiling compounds.
1.4 Sensitivity.
The sensitivity limit for a compound is defined as the minimum detectable concentration of that compound, or the concentration that produces a signal-to-noise ratio of three to one. The minimum detectable concentration is determined during the presurvey calibration for each compound.
2.0 Summary of Method.
The major organic components of a gas mixture are separated by gas chromatography (GC) and individually quantified by flame ionization, photoionization, electron capture, or other appropriate detection principles. The retention times of each separated component are compared with those of known compounds under identical conditions. Therefore, the analyst confirms the identity and approximate concentrations of the organic emission components beforehand. With this information, the analyst then prepares or purchases commercially available standard mixtures to calibrate the GC under conditions identical to those of the samples. The analyst also determines the need for sample dilution to avoid detector saturation, gas stream filtration to eliminate particulate matter, and prevention of moisture condensation.
3.0 Definitions. [Reserved]
4.0 Interferences.
4.1 Resolution interferences that may occur can be eliminated by appropriate GC column and detector choice or by shifting the retention times through changes in the column flow rate and the use of temperature programming.
4.2 The analytical system is demonstrated to be essentially free from contaminants by periodically analyzing blanks that consist of hydrocarbon-free air or nitrogen.
4.3 Sample cross-contamination that occurs when high-level and low-level samples or standards are analyzed alternately is best dealt with by thorough purging of the GC sample loop between samples.
4.4 To assure consistent detector response, calibration gases are contained in dry air. To adjust gaseous organic concentrations when water vapor is present in the sample, water vapor concentrations are determined for those samples, and a correction factor is applied.
4.5 The gas chromatograph run time must be sufficient to clear all eluting peaks from the column before proceeding to the next run (in order to prevent sample carryover).
5.0 Safety.
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method. The analyzer users manual should be consulted for specific precautions to be taken with regard to the analytical procedure.
6.0 Equipment and Supplies.
6.1 equipment needed for the presurvey sampling procedure can be found in Section 16.1.1.
6.2 equipment needed for the integrated bag sampling and analysis procedure can be found in Section 8.2.1.1.1.
6.3 equipment needed for direct interface sampling and analysis can be found in Section 8.2.2.1.
6.4 equipment needed for the dilution interface sampling and analysis can be found in Section 8.2.3.1.
6.5 equipment needed for adsorbent tube sampling and analysis can be found in Section 8.2.4.1.
7.0 Reagents and Standards.
7.1 Reagents needed for the presurvey sampling procedure can be found in Section 16.1.2.
7.2 Quality Assurance Audit Samples. When making compliance determinations, and upon availability, an audit sample may be obtained from the appropriate EPA Regional Office or from the responsible enforcement authority.
NOTE: The responsible enforcement authority should be notified at least 30 days prior to the test date to allow sufficient time for sample delivery.
8.0 Sample Collection, Preservation, Storage, and Transport.
8.2 Final Sampling and Analysis Procedure.
Considering safety (flame hazards) and the source conditions, select an appropriate sampling and analysis procedure (Section 8.2.1, 8.2.2, 8.2.3 or 8.2.4). In situations where a hydrogen flame is a hazard and no intrinsically safe GC is suitable, use the flexible bag collection technique or an adsorption technique.
8.2.1 Integrated Bag Sampling and Analysis.
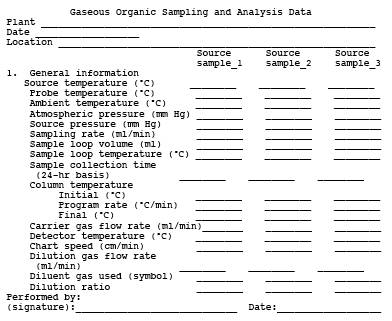
8.2.1.1 Evacuated Container Sampling Procedure. In this procedure, the bags are filled by evacuating the rigid air-tight container holding the bags. Use a field sample data sheet as shown in Figure 18-10. Collect triplicate samples from each sample location.
8.2.1.1.1 Apparatus.
8.2.1.1.1.1 Probe. Stainless steel, Pyrex glass, or Teflon tubing Probe, according to the duct temperature, with Teflon tubing of sufficient length to connect to the sample bag. Use stainless steel or Teflon unions to connect Probe and sample line.
8.2.1.1.1.2 Quick Connects. Male (2) and female (2) of stainless steel construction.
8.2.1.1.1.3 Needle Valve. To control gas flow.
8.2.1.1.1.4 pump. Leakless Teflon-coated diaphragm-type pump or equivalent. To deliver at least 1 liter/min.
8.2.1.1.1.5 Charcoal Adsorption Tube. Tube filled with activated charcoal, with glass wool plugs at each end, to adsorb organic vapors.
8.2.1.1.1.6 flowmeter. O to 500-ml flow range; with manufacturer's calibration curve.
8.2.1.1.2 Sampling Procedure. To obtain a sample, assemble the sample train as shown in Figure 18-9. Leak-check both the bag and the container. Connect the vacuum line from the needle valve to the Teflon sample line from the Probe. Place the end of the Probe at the centroid of the stack or at a point no closer to the walls than 1 m, and start the pump. Set the flow rate so that the final volume of the sample is approximately 80 percent of the bag capacity. After allowing sufficient time to purge the line several times, connect the vacuum line to the bag, and evacuate until the rotameter indicates no flow. Then position the sample and vacuum lines for sampling, and begin the actual sampling, keeping the rate proportional to the stack velocity. As a precaution, direct the gas exiting the rotameter away from sampling personnel. At the end of the sample period, shut off the pump, disconnect the sample line from the bag, and disconnect the vacuum line from the bag container. Record the source temperature, barometric pressure, ambient temperature, sampling flow rate, and initial and final sampling time on the data sheet shown in Figure 18-10. Protect the Tedlar Bag and its container from sunlight. Record the time lapsed between sample collection and analysis, and then conduct the recovery procedure in Section 8.4.2.
8.2.1.2 Direct pump Sampling Procedure. Follow 8.2.1.1, except place the pump and needle valve between the Probe and the bag. Use a pump and needle valve constructed of inert material not affected by the stack gas. Leak-check the system, and then purge with stack gas before connecting to the previously evacuated bag.
8.2.1.3 Explosion Risk Area Bag Sampling Procedure. Follow 8.2.1.1 except replace the pump with another evacuated can (see Figure 18-9a). Use this method whenever there is a possibility of an explosion due to pumps, heated Probes, or other flame producing equipment.
8.2.1.4 Other Modified Bag Sampling Procedures. In the event that condensation is observed in the bag while collecting the sample and a direct interface system cannot be used, heat the bag during collection, and maintain it at a suitably elevated temperature during all subsequent operations. (NOTE: Take care to leak-check the system prior to the dilutions so as not to create a potentially explosive atmosphere.) As an alternative, collect the sample gas, and simultaneously dilute it in the Tedlar Bag.
8.2.1.4.1 First Alternative Procedure. Heat the box containing the sample bag to 120 C (± 5 C). Then transport the bag as rapidly as possible to the analytical area while maintaining the heating, or cover the box with an insulating blanket. In the analytical area, keep the box heated to 120 C (±5 C) until analysis. Be sure that the method of heating the box and the control for the heating circuit are compatible with the safety restrictions required in each area.
8.2.1.4.2 Second Alternative Procedure. Prefill the Tedlar Bag with a known quantity of inert gas. meter the inert gas into the bag according to the procedure for the preparation of gas concentration standards of volatile liquid materials (Section 10.1.2.2), but eliminate the midget impinger section. Take the partly filled bag to the source, and meter the source gas into the bag through heated sampling lines and a heated flowmeter, or Teflon positive displacement pump. Verify the dilution factors before sampling each bag through dilution and analysis of gases of known concentration.
8.2.1.5 Analysis of Bag Samples.
8.2.1.5.1 Apparatus. Same as Section 8.1. A minimum of three gas standards are required.
8.2.1.5.2 Procedure.
8.2.1.5.2.1 Establish proper GC operating conditions as described in Section 10.2, and record all data listed in Figure 18-7. Prepare the GC so that gas can be drawn through the sample valve. Flush the sample loop with calibration gas mixture, and activate the valve (sample pressure at the inlet to the GC introduction valve should be similar during calibration as during actual sample analysis). Obtain at least three chromatograms for the mixture. The results are acceptable when the peak areas for the three injections agree to within 5 percent of their average. If they do not agree, run additional samples or correct the analytical techniques until this requirement is met. Then analyze the other two calibration mixtures in the same manner. Prepare a calibration curve as described in Section 10.2.
8.2.1.5.2.2 Analyze the two field audit samples as described in Section 9.2 by connecting each Tedlar Bag containing an audit gas mixture to the sampling valve. Calculate the results; record and report the data to the audit supervisor.
8.2.1.5.2.3 Analyze the three source gas samples by connecting each bag to the sampling valve with a piece of Teflon tubing identified with that bag. Analyze each bag sample three times. Record the data in Figure 18-11. If certain items do not apply, use the notation "N.A." If the bag has been maintained at an elevated temperature as described in Section 8.2.1.4, determine the stack gas water content by Method 4. After all samples have been analyzed, repeat the analysis of the mid-level calibration gas for each compound. Compare the average response factor of the pre- and post-test analysis for each compound. If they differ by > 5 percent, analyze the other calibration gas levels for that compound, and prepare a calibration curve using all the pre- and post-test calibration gas mixture values. If the two response factor averages (pre- and post-test) differ by less than 5 percent from their mean value, the tester has the option of using only the pre-test calibration curve to generate the concentration values.
8.2.1.6 Determination of Bag Water Vapor Content. Measure the ambient temperature and barometric pressure near the bag. From a water saturation vapor pressure table, determine and record the water vapor content of the bag as a decimal figure. (Assume the relative humidity to be 100 percent unless a lesser value is known.) If the bag has been maintained at an elevated temperature as described in Section 8.2.1.4, determine the stack gas water content by Method 4.
8.2.1.7 Audit Gas Analysis. Immediately prior to the analysis of the stack gas samples, perform audit analyses as described in Section 9.2.
8.2.1.8 Emission Calculations. From the calibration curve described in Section 8.2.1.5, select the value of Cs that corresponds to the peak area. Calculate the concentration Cc in ppm, dry basis, of each organic in the sample using Equation 18-5 in Section 12.6.
8.2.2 Direct Interface Sampling and Analysis Procedure.
The direct interface procedure can be used provided that the moisture content of the gas does not interfere with the analysis procedure, the physical requirements of the equipment can be met at the site, and the source gas concentration falls within the linear range of the detector. Adhere to all safety requirements with this method.
8.2.2.1 Apparatus.
8.2.2.1.1 Probe. Constructed of stainless steel, Pyrex glass, or Teflon tubing as dictated by duct temperature and reactivity of target compounds. A filter or glass wool plug may be needed if particulate is present in the stack gas. If necessary, heat the Probe with heating tape or a special heating unit capable of maintaining a temperature greater than 110C.
8.2.2.1.2 Sample Lines. 6.4-mm OD (or other diameter as needed) Teflon lines, heat-traced to prevent condensation of material (greater than 110C).
8.2.2.1.3 Quick Connects. To connect sample line to gas sampling valve on GC instrument and to pump unit used to withdraw source gas. Use a quick connect or equivalent on the cylinder or bag containing calibration gas to allow connection of the calibration gas to the gas sampling valve.
8.2.2.1.4 thermocouple Readout Device. Potentiometer or digital thermometer, to measure source temperature and Probe temperature.
8.2.2.1.5 Heated Gas Sampling Valve. Of two-position, six-port design, to allow sample loop to be purged with source gas or to direct source gas into the GC instrument.
8.2.2.1.6 Needle Valve. To control gas sampling rate from the source.
8.2.2.1.7 pump. Leakless Teflon-coated diaphragm-type pump or equivalent, capable of at least 1 liter/minute sampling rate.
8.2.2.1.8 flowmeter. Of suitable range to measure sampling rate.
8.2.2.1.9 Charcoal Adsorber. To adsorb organic vapor vented from the source to prevent exposure of personnel to source gas.
8.2.2.1.10 Gas Cylinders. Carrier gas, oxygen and fuel as needed to run GC and detector.
8.2.2.1.11 Gas Chromatograph. Capable of being moved into the field, with detector, heated gas sampling valve, column required to complete separation of desired components, and option for temperature programming.
8.2.2.1.12 Recorder/Integrator. To record results.
8.2.2.2 Procedure. Calibrate the GC using the procedures in Section 8.2.1.5.2.1. To obtain a stack gas sample, assemble the sampling system as shown in Figure 18-12. Make sure all connections are tight. Turn on the Probe and sample line heaters. As the temperature of the Probe and heated line approaches the target temperature as indicated on the thermocouple readout device, control the heating to maintain a temperature greater than 110C. Conduct a 3-point calibration of the GC by analyzing each gas mixture in triplicate. Generate a calibration curve. Place the inlet of the Probe at the centroid of the duct, or at a point no closer to the walls than 1 m, and draw source gas into the Probe, heated line, and sample loop. After thorough flushing, analyze the stack gas sample using the same conditions as for the calibration gas mixture. For each run, sample, analyze, and record five consecutive samples. A test consists of three runs (five samples per run times three runs, for a total of fifteen samples). After all samples have been analyzed, repeat the analysis of the mid-level calibration gas for each compound. For each calibration standard, compare the pre- and post-test average response factors (RF) for each compound. If the two calibration RF values (pre and post-analysis) differ by more than 5 percent from their mean value, then analyze the other calibration gas levels for that compound and determine the stack gas sample concentrations by comparison to both calibration curves (this is done by preparing a calibration curve using all the pre and post-test calibration gas mixture values). If the two calibration RF values differ by less than 5 percent from their mean value, the tester has the option of using only the pre-test calibration curve to generate the concentration values. Record this calibration data and the other required data on the data sheet shown in Figure 18-11, deleting the dilution gas information. (NOTE: Take care to draw all samples, calibration mixtures, and audits through the sample loop at the same pressure.)
8.2.2.3 Determination of Stack Gas Moisture Content. Use Method 4 to measure the stack gas moisture content.
8.2.2.4 Quality Assurance. Same as Section 8.2.1.7. Introduce the audit gases in the sample line immediately following the Probe.
8.2.2.5 Emission Calculations. Same as Section 8.2.1.8.
8.2.3 Dilution Interface Sampling and Analysis Procedure.
Source samples that contain a high concentration of organic materials may require dilution prior to analysis to prevent saturating the GC detector. The apparatus required for this direct interface procedure is basically the same as that described in the Section 8.2.2, except a dilution system is added between the heated sample line and the gas sampling valve. The apparatus is arranged so that either a 10:1 or 100:1 dilution of the source gas can be directed to the chromatograph. A pump of larger capacity is also required, and this pump must be heated and placed in the system between the sample line and the dilution apparatus.
8.2.3.1 Apparatus. The equipment required in addition to that specified for the direct interface system is as follows:
8.2.3.1.1 sample pump>. Leakless Teflon-coated diaphragm-type that can withstand being heated to 120C and deliver 1.5 liters/minute.
8.2.3.1.2 Dilution pumps. Two Model A-150 Komhyr Teflon positive displacement type delivering 150 cc/minute, or equivalent. As an option, calibrated flowmeters can be used in conjunction with Teflon-coated diaphragm pumps.
8.2.3.1.3 Valves. Two Teflon three-way valves, suitable for connecting to Teflon tubing.
8.2.3.1.4 flowmeters. Two, for measurement of diluent gas.
8.2.3.1.5 Diluent Gas with Cylinders and Regulators. Gas can be nitrogen or clean dry air, depending on the nature of the source gases.
8.2.3.1.6 Heated Box. Suitable for being heated to 120C, to contain the three pumps, three-way valves, and associated connections. The box should be equipped with quick connect fittings to facilitate connection of: (1) the heated sample line from the Probe, (2) the gas sampling valve, (3) the calibration gas mixtures, and (4) diluent gas lines. A schematic diagram of the components and connections is shown in Figure 18-13. The heated box shown in Figure 18-13 is designed to receive a heated line from the Probe. An optional design is to build a Probe unit that attaches directly to the heated box. In this way, the heated box contains the controls for the Probe heaters, or, if the box is placed against the duct being sampled, it may be possible to eliminate the Probe heaters. In either case, a heated Teflon line is used to connect the heated box to the gas sampling valve on the chromatograph.
NOTE: Care must be taken to leak-check the system prior to the dilutions so as not to create a potentially explosive atmosphere.
8.2.3.2 Procedure.
8.2.3.2.1 Assemble the apparatus by connecting the heated box, shown in Figure 18-13, between the heated sample line from the Probe and the gas sampling valve on the chromatograph. Vent the source gas from the gas sampling valve directly to the charcoal filter, eliminating the pump and rotameter. Heat the sample Probe, sample line, and heated box. Insert the Probe and source thermocouple at the centroid of the duct, or to a point no closer to the walls than 1 m. Measure the source temperature, and adjust all heating units to a temperature 0 to 3C above this temperature. If this temperature is above the safe operating temperature of the Teflon components, adjust the heating to maintain a temperature high enough to prevent condensation of water and organic compounds (greater than 110C). Calibrate the GC through the dilution system by following the procedures in Section 8.2.1.5.2.1. Determine the concentration of the diluted calibration gas using the dilution factor and the certified concentration of the calibration gas. Record the pertinent data on the data sheet shown in Figure 18-11.
8.2.3.2.2 Once the dilution system and GC operations are satisfactory, proceed with the analysis of source gas, maintaining the same dilution settings as used for the standards.
8.2.3.2.3 Analyze the audit samples using either the dilution system, or directly connect to the gas sampling valve as required. Record all data and report the results to the audit supervisor.
8.2.3.3 Determination of Stack Gas Moisture Content. Same as Section 8.2.2.3.
8.2.3.4 Quality Assurance. Same as Section 8.2.2.4.
8.2.3.5 Emission Calculations. Same as section 8.2.2.5, with the dilution factor applied.
8.2.4 Adsorption Tube Procedure.
Any commercially available adsorbent is allowed for the purposes of this method, as long as the recovery study criteria in Section 8.4.3 are met. Help in choosing the adsorbent may be found by calling the distributor, or the tester may refer to National Institute for Occupational Safety and Health (NIOSH) methods for the particular organics to be sampled. For some adsorbents, the principal interferent will be water vapor. If water vapor is thought to be a problem, the tester may place a midget impinger in an ice bath before the adsorbent tubes. If this option is chosen, the water catch in the midget impinger shall be analyzed for the target compounds. Also, the spike for the recovery study (in Section 8.4.3) shall be conducted in both the midget impinger and the adsorbent tubes. The combined recovery (add the recovered amount in the impinger and the adsorbent tubes to calculate R) shall then meet the criteria in Section 8.4.3. NOTE: Post-test leak-checks are not allowed for this technique since this can result in sample contamination.
8.2.4.1 Additional Apparatus. The following items (or equivalent) are suggested.
8.2.4.1.1 Probe. Borosilicate glass or stainless steel, approximately 6-mm ID, with a heating system if water condensation is a problem, and a filter (either in-stack or out-of-stack, heated to stack temperature) to remove particulate matter. In most instances, a plug of glass wool is a satisfactory filter.
8.2.4.1.2 Flexible tubing. To connect Probe to adsorption tubes. Use a material that exhibits minimal sample adsorption.
8.2.4.1.3 Leakless sample pump>. flow controlled, constant rate pump, with a set of limiting (sonic) orifices.
8.2.4.1.4 Bubble-Tube flowmeter. Volume accuracy within 1 percent, to calibrate pump.
8.2.4.1.5 Stopwatch. To time sampling and pump rate calibration.
8.2.4.1.6 Adsorption Tubes. Precleaned adsorbent, with mass of adsorbent to be determined by calculating breakthrough volume and expected concentration in the stack.
8.2.4.1.7 barometer. Accurate to 5 mm Hg, to measure atmospheric pressure during sampling and pump calibration.
8.2.4.1.8 Rotameter. O to 100 cc/min, to detect changes in flow rate during sampling.
8.2.4.2 Sampling and Analysis.
8.2.4.2.1 Calibrate the pump and limiting orifice flow rate through adsorption tubes with the bubble tube flowmeter before sampling. The sample system can be operated as a "recirculating loop" for this operation. Record the ambient temperature and barometric pressure. Then, during sampling, use the rotameter to verify that the pump and orifice sampling rate remains constant.
8.2.4.2.2 Use a sample Probe, if required, to obtain the sample at the centroid of the duct, or at a point no closer to the walls than 1 m. Minimize the length of flexible tubing between the Probe and adsorption tubes. Several adsorption tubes can be connected in series, if the extra adsorptive capacity is needed. Adsorption tubes should be maintained vertically during the test in order to prevent channeling. Provide the gas sample to the sample system at a pressure sufficient for the limiting orifice to function as a sonic orifice. Record the total time and sample flow rate (or the number of pump strokes), the barometric pressure, and ambient temperature. Obtain a total sample volume commensurate with the expected concentration(s) of the volatile organic(s) present, and recommended sample loading factors (weight sample per weight adsorption media). Laboratory tests prior to actual sampling may be necessary to predetermine this volume. If water vapor is present in the sample at concentrations above 2 to 3 percent, the adsorptive capacity may be severely reduced. Operate the gas chromatograph according to the manufacturer's instructions. After establishing optimum conditions, verify and document these conditions during all operations. Calibrate the instrument. Analyze the audit samples (see Section 16.1.4.3), then the emission samples.
8.2.4.3 Standards and calibration. If using thermal desorption, obtain calibration gases using the procedures in Section 10.1. If using solvent extraction, prepare liquid standards in the desorption solvent. Use a minimum of three different standards; select the concentrations to bracket the expected average sample concentration. Perform the calibration before and after each day's sample analyses using the procedures in Section 8.2.1.5.2.1.
8.2.4.4 Quality Assurance.
8.2.4.4.1 Determine the recovery efficiency of the pollutants of interest according to Section 8.4.3.
8.2.4.4.2 Determination of Sample Collection Efficiency (Optional). If sample breakthrough is thought to be a problem, a routine procedure for determining breakthrough is to analyze the primary and backup portions of the adsorption tubes separately. If the backup portion exceeds 10 percent of the total amount (primary and back-up), it is usually a sign of sample breakthrough. For the purposes of this method, only the recovery efficiency value (Section 8.4.3) is used to determine the appropriateness of the sampling and analytical procedure.
8.2.4.4.3 Volume flow Rate Checks. Perform this check immediately after sampling with all sampling train components in place. Use the bubble-tube flowmeter to measure the pump volume flow rate with the orifice used in the test sampling, and record the result. If it has changed by more than 5 but less than 20 percent, calculate an average flow rate for the test. If the flow rate has changed by more than 20 percent, recalibrate the pump and repeat the sampling.
8.2.4.4.4 Calculations. Correct all sample volumes to standard conditions. If a sample dilution system has been used, multiply the results by the appropriate dilution ratio. Correct all results according to the applicable procedure in Section 8.4.3. Report results as ppm by volume, dry basis.
8.3 Reporting of Results.
At the completion of the field analysis portion of the study, ensure that the data sheets shown in Figure 18-11 have been completed. Summarize this data on the data sheets shown in Figure 18-15.
8.4 Recovery Study.
After conducting the presurvey and identifying all of the pollutants of interest, conduct the appropriate recovery study during the test based on the sampling system chosen for the compounds of interest.
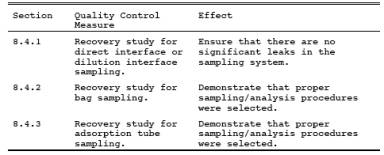
8.4.1 Recovery Study for Direct Interface or Dilution Interface Sampling.
If the procedures in Section 8.2.2 or 8.2.3 are to be used to analyze the stack gas, conduct the calibration procedure as stated in Section 8.2.2.2 or 8.2.3.2, as appropriate. Upon successful completion of the appropriate calibration procedure, attach the mid-level calibration gas for at least one target compound to the inlet of the Probe or as close as possible to the inlet of the Probe, but before the filter. Repeat the calibration procedure by sampling and analyzing the mid-level calibration gas through the entire sampling and analytical system in triplicate. The mean of the calibration gas response sampled through the Probe shall be within 10 percent of the analyzer response. If the difference in the two means is greater than 10 percent, check for leaks throughout the sampling system and repeat the analysis of the standard through the sampling system until this criterion is met.
8.4.2 Recovery Study for Bag Sampling.
8.4.2.1 Follow the procedures for the bag sampling and analysis in Section 8.2.1. After analyzing all three bag samples, choose one of the bag samples and tag this bag as the spiked bag. Spike the chosen bag sample with a known mixture (gaseous or liquid) of all of the target pollutants. The theoretical concentration, in ppm, of each spiked compound in the bag shall be 40 to 60 percent of the average concentration measured in the three bag samples. If a target compound was not detected in the bag samples, the concentration of that compound to be spiked shall be 5 times the limit of detection for that compound. Store the spiked bag for the same period of time as the bag samples collected in the field. After the appropriate storage time has passed, analyze the spiked bag three times. Calculate the average fraction recovered (R) of each spiked target compound with the equation in Section 12.7.
8.4.2.2 For the bag sampling technique to be considered valid for a compound, 0.70 ≤R ≤1.30. If the R value does not meet this criterion for a target compound, the sampling technique is not acceptable for that compound, and therefore another sampling technique shall be evaluated for acceptance (by repeating the recovery study with another sampling technique). Report the R value in the test report and correct all field measurements with the calculated R value for that compound by using the equation in Section 12.8.
8.4.3 Recovery Study for Adsorption Tube Sampling.
If following the adsorption tube procedure in Section 8.2.4, conduct a recovery study of the compounds of interest during the actual field test. Set up two identical sampling trains. Collocate the two sampling Probes in the stack. The Probes shall be placed in the same horizontal plane, where the first Probe tip is 2.5 cm from the outside edge of the other. One of the sampling trains shall be designated the spiked train and the other the unspiked train. Spike all of the compounds of interest (in gaseous or liquid form) onto the adsorbent tube(s) in the spiked train before sampling. The mass of each spiked compound shall be 40 to 60 percent of the mass expected to be collected with the unspiked train. Sample the stack gas into the two trains simultaneously. Analyze the adsorbents from the two trains utilizing identical analytical procedures and instrumentation. Determine the fraction of spiked compound recovered (R) using the equations in Section 12.9.
8.4.3.1 Repeat the procedure in Section 8.4.3 twice more, for a total of three runs. In order for the adsorbent tube sampling and analytical procedure to be acceptable for a compound, 0.70 ≤R ≤1.30 (R in this case is the average of three runs). If the average R value does not meet this criterion for a target compound, the sampling technique is not acceptable for that compound, and therefore another sampling technique shall be evaluated for acceptance (by repeating the recovery study with another sampling technique). Report the R value in the test report and correct all field measurements with the calculated R value for that compound by using the equation in Section 12.8.
9.0 Quality Control.
9.1 Miscellaneous Quality Control Measures
9.2 Quality Assurance for Laboratory Procedures.
Immediately after the preparation of the calibration curves, the analysis audit described in 40 CFR Part 61, Appendix C, Procedure 2: "Procedure for Field Auditing GC Analysis," should be performed if audit materials are available. The information required to document the analysis of the audit samples has been included on the example data sheets shown in Figures 18-3 and 18-7. The audit analyses should agree with the certified audit concentrations within 10 percent. Audit sample results shall be submitted according to directions provided with the audit samples.
10.0 Calibration and Standardization.
10.1 calibration Standards.
Obtain calibration gas standards for each target compound to be analyzed. Commercial cylinder gases certified by the manufacturer to be accurate to within 1 percent of the certified label value are preferable, although cylinder gases certified by the manufacturer to 2 percent accuracy are allowed. Another option allowed by this method is for the tester to obtain high concentration certified cylinder gases and then use a dilution system meeting the requirements of Test Method 205, 40 CFR Part 51, Appendix M to make multi-level calibration gas standards. Prepare or obtain enough calibration standards so that there are three different concentrations of each organic compound expected to be measured in the source sample. For each organic compound, select those concentrations that bracket the concentrations expected in the source samples. A calibration standard may contain more than one organic compound. If samples are collected in adsorbent tubes and extracted using solvent extraction, prepare or obtain standards in the same solvent used for the sample extraction procedure. Verify the stability of all standards for the time periods they are used.
10.2 Preparation of calibration Curves.
10.2.1 Establish proper GC conditions, then flush the sampling loop for 30 seconds. Allow the sample loop pressure to equilibrate to atmospheric pressure, and activate the injection valve. Record the standard concentration, attenuator factor, injection time, chart speed, retention time, peak area, sample loop temperature, column temperature, and carrier gas flow rate. Analyze each standard in triplicate.
10.2.2 Repeat this procedure for each standard. Prepare a graphical plot of concentration (Cs) versus the calibration area values. Perform a regression analysis, and draw the least square line.
11.0 Analytical Procedures.
11.1 Analysis Development.
11.1.1 Selection of GC Parameters.
11.1.1.1 Column Choice. Based on the initial contact with plant personnel concerning the plant process and the anticipated emissions, choose a column that provides good resolution and rapid analysis time. The choice of an appropriate column can be aided by a literature search, contact with manufacturers of GC columns, and discussion with personnel at the emission source.
NOTE: Most column manufacturers keep excellent records on their products. Their technical service departments may be able to recommend appropriate columns and detector type for separating the anticipated compounds, and they may be able to provide information on interferences, optimum operating conditions, and column limitations. Plants with analytical laboratories may be able to provide information on their analytical procedures.
11.1.1.2 Preliminary GC Adjustment. Using the standards and column obtained in Section 11.1.1.1, perform initial tests to determine appropriate GC conditions that provide good resolution and minimum analysis time for the compounds of interest.
11.1.1.3 Preparation of Presurvey Samples. If the samples were collected on an adsorbent, extract the sample as recommended by the manufacturer for removal of the compounds with a solvent suitable to the type of GC analysis. Prepare other samples in an appropriate manner.
11.1.1.4 Presurvey Sample Analysis.
11.1.1.4.1 Before analysis, heat the presurvey sample to the duct temperature to vaporize any condensed material. Analyze the samples by the GC procedure, and compare the retention times against those of the calibration samples that contain the components expected to be in the stream. If any compounds cannot be identified with certainty by this procedure, identify them by other means such as GC/mass spectroscopy (GC/MS) or GC/infrared techniques. A GC/MS system is recommended.
11.1.1.4.2 Use the GC conditions determined by the procedure of Section 11.1.1.2 for the first injection. Vary the GC parameters during subsequent injections to determine the optimum settings. Once the optimum settings have been determined, perform repeat injections of the sample to determine the retention time of each compound. To inject a sample, draw sample through the loop at a constant rate (100 ml/min for 30 seconds). Be careful not to pressurize the gas in the loop. Turn off the pump and allow the gas in the sample loop to come to ambient pressure. Activate the sample valve, and record injection time, loop temperature, column temperature, carrier flow rate, chart speed, and attenuator setting. Calculate the retention time of each peak using the distance from injection to the peak maximum divided by the chart speed. Retention times should be repeatable within O.5 seconds.
11.1.1.4.3 If the concentrations are too high for appropriate detector response, a smaller sample loop or dilutions may be used for gas samples, and, for liquid samples, dilution with solvent is appropriate. Use the standard curves (Section 10.2) to obtain an estimate of the concentrations.
11.1.1.4.4 Identify all peaks by comparing the known retention times of compounds expected to be in the retention times of peaks in the sample. Identify any remaining unidentified peaks which have areas larger than 5 percent of the total using a GC/MS, or estimation of possible compounds by their retention times compared to known compounds, with confirmation by further GC analysis.
12.0 Data Analysis and Calculations.
12.1 Nomenclature.
Bws = Water vapor content of the bag sample or stack gas, proportion by volume.
Cs = Concentration of the organic from the calibration curve, ppm.
Gv = Gas volume or organic compound injected, ml.
Lv = Liquid volume of organic injected,l.
M = Molecular weight of organic, g/g-mole.
ms = Total mass of compound measured on adsorbent with spiked train (g).
mu = Total mass of compound measured on adsorbent with unspiked train (g).
mv = Mass per volume of spiked compound measured (g/L).
Pi = Barometric or absolute sample loop pressure at time of sample analysis, mm Hg.
Pm = Absolute pressure of dry gas meter, mm Hg.
Pr = Reference pressure, the barometric pressure or absolute sample loop pressure recorded during calibration, mm Hg.
Ps = Absolute pressure of syringe before injection, mm Hg.
qc = flow rate of the calibration gas to be diluted.
qc1 = flow rate of the calibration gas to be diluted in stage 1.
qc2 = flow rate of the calibration gas to be diluted in stage 2.
qd = Diluent gas flow rate.
qd1 = flow rate of diluent gas in stage 1.
qd2 = flow rate of diluent gas in stage 2.
s = Theoretical concentration (ppm) of spiked target compound in the bag.
S = Theoretical mass of compound spiked onto adsorbent in spiked train (g).
t = Measured average concentration (ppm) of target compound and source sample (analysis results subsequent to bag spiking)
Ti = Sample loop temperature at the time of sample analysis, K.
Tm = Absolute temperature of dry gas meter, K.
Ts = Absolute temperature of syringe before injection, K.
u = Source sample average concentration (ppm) of target compound in the bag (analysis results before bag spiking).
Vm = Gas volume indicated by dry gas meter, liters.
vs = volume of stack gas sampled with spiked train (L).
vu = volume of stack gas sampled with unspiked train (L).
X = Mole or volume fraction of the organic in the calibration gas to be diluted.
Y = dry gas meter calibration factor, dimensionless.
l = Liquid organic density as determined, g/ml.
24.055 = Ideal gas molar volume at 293 K and 760 mm Hg, liters/g-mole.
1000 = Conversion factor, ml/liter.
106 = Conversion to ppm.
12.2 Calculate the concentration, Cs, in ppm using the following equation:

12.3 Calculate the concentration, Cs, in ppm of the organic in the final gas mixture using the following equation:
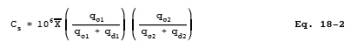
12.4 Calculate each organic standard concentration, Cs, in ppm using the following equation:

12.5 Calculate each organic standard concentration, Cs, in ppm using the following equation:
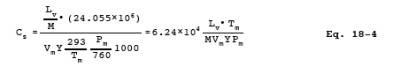
12.6 Calculate the concentration, Cc, in ppm, dry basis, of each organic is the sample using the following equation:

12.7 Calculate the average fraction recovered (R) of each spiked target compound using the following equation:

12.8 Correct all field measurements with the calculated R value for that compound using the following equation:

12.9 Determine the mass per volume of spiked compound measured using the following equation:

12.10 Calculate the fraction of spiked compound recovered, R, using the following equation:

13.0 Method Performance.
13.1 Since a potential sample may contain a variety of compounds from various sources, a specific precision limit for the analysis of field samples is impractical. Precision in the range of 5 to 10 percent relative standard deviation (RSD) is typical for gas chromatographic techniques, but an experienced GC operator with a reliable instrument can readily achieve 5 percent RSD. For this method, the following combined GC/operator values are required.
(a) Precision. Triplicate analyses of calibration standards fall within 5 percent of their mean value. (b) Accuracy. Analysis results of prepared audit samples are within 10 percent of preparation values. (c) Recovery. After developing an appropriate sampling and analytical system for the pollutants of interest, conduct the procedure in Section 8.4. Conduct the appropriate recovery study in Section 8.4 at each sampling point where the method is being applied. Submit the data and results of the recovery procedure with the reporting of results under Section 8.3.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 Alternative Procedures.
16.1 Optional Presurvey and Presurvey Sampling.
NOTE: Presurvey screening is optional. Presurvey sampling should be conducted for sources where the target pollutants are not known from previous tests and/or process knowledge. Perform a presurvey for each source to be tested. Refer to Figure 18-1. Some of the information can be collected from literature surveys and source personnel. Collect gas samples that can be analyzed to confirm the identities and approximate concentrations of the organic emissions.
16.1.1 Apparatus.
This apparatus list also applies to Sections 8.2 and 11.
16.1.1.1 Teflon tubing. (Mention of trade names or specific products does not constitute endorsement by the U.S. Environmental Protection Agency.) Diameter and length determined by connection requirements of cylinder regulators and the GC. Additional tubing is necessary to connect the GC sample loop to the sample.
16.1.1.2 Gas Chromatograph. GC with suitable detector, columns, temperature-controlled sample loop and valve assembly, and temperature programmable oven, if necessary. The GC shall achieve sensitivity requirements for the compounds under study.
16.1.1.3 pump. Capable of pumping 100 ml/min. For flushing sample loop.
16.1.1.4 flow meter. To measure flow rates.
16.1.1.5 Regulators. Used on gas cylinders for GC and for cylinder standards.
16.1.1.6 Recorder. Recorder with linear strip chart is minimum acceptable. Integrator (optional) is recommended.
16.1.1.7 Syringes. 0.5-ml, 1.O- and 10-microliter size, calibrated, maximum accuracy (gas tight) for preparing calibration standards. Other appropriate sizes can be used.
16.1.1.8 tubing Fittings. To plumb GC and gas cylinders.
16.1.1.9 Septa. For syringe injections.
16.1.1.10 glass Jars. If necessary, clean, colored glass jars with Teflon-lined lids for condensate sample collection. Size depends on volume of condensate.
16.1.1.11 Soap Film flowmeter. To determine flow rates.
16.1.1.12 Tedlar Bags. 10- and 50-liter capacity, for preparation of standards.
16.1.1.13 dry gas meter with temperature and Pressure gauges. Accurate to ± 2 percent, for preparation of gas standards.
16.1.1.14 Midget impinger/Hot Plate Assembly. For preparation of gas standards.
16.1.1.15 Sample Flasks. For presurvey samples, must have gas-tight seals.
16.1.1.16 Adsorption Tubes. If necessary, blank tubes filled with necessary adsorbent (charcoal, Tenax, XAD-2, etc.) for presurvey samples.
16.1.1.17 Personnel Sampling pump. Calibrated, for collecting adsorbent tube presurvey samples.
16.1.1.18 Dilution System. Calibrated, the dilution system is to be constructed following the specifications of an acceptable method.
16.1.1.19 Sample Probes. Pyrex or stainless steel, of sufficient length to reach centroid of stack, or a point no closer to the walls than 1 m.
16.1.1.20 barometer. To measure barometric pressure.
16.1.2 Reagents.
16.1.2.1 Water. Deionized distilled.
16.1.2.2 Methylene chloride.
16.1.2.3 calibration Gases. A series of standards prepared for every compound of interest.
16.1.2.4 Organic Compound Solutions. Pure (99.9 percent), or as pure as can reasonably be obtained, liquid samples of all the organic compounds needed to prepare calibration standards.
16.1.2.5 Extraction Solvents. For extraction of adsorbent tube samples in preparation for analysis.
16.1.2.6 Fuel. As recommended by the manufacturer for operation of the GC.
16.1.2.7 Carrier Gas. Hydrocarbon free, as recommended by the manufacturer for operation of the detector and compatibility with the column.
16.1.2.8 Zero Gas. Hydrocarbon free air or nitrogen, to be used for dilutions, blank preparation, and standard preparation.
16.1.3 Sampling.
16.1.3.1 Collection of Samples with glass Sampling Flasks. Presurvey samples may be collected in precleaned 250-ml double-ended glass sampling flasks. Teflon stopcocks, without grease, are preferred. Flasks should be cleaned as follows: Remove the stopcocks from both ends of the flasks, and wipe the parts to remove any grease. Clean the stopcocks, barrels, and receivers with methylene chloride (or other non-target pollutant solvent, or heat and humidified air). Clean all glass ports with a soap solution, then rinse with tap and deionized distilled water. Place the flask in a cool glass annealing furnace, and apply heat up to 500C. Maintain at this temperature for 1 hours. After this time period, shut off and open the furnace to allow the flask to cool. Return the stopcocks to the flask receivers. Purge the assembly with high-purity nitrogen for 2 to 5 minutes. Close off the stopcocks after purging to maintain a slight positive nitrogen pressure. Secure the stopcocks with tape. Presurvey samples can be obtained either by drawing the gases into the previously evacuated flask or by drawing the gases into and purging the flask with a rubber suction bulb.
16.1.3.1.1 Evacuated Flask Procedure. Use a high-vacuum pump to evacuate the flask to the capacity of the pump; then close off the stopcock leading to the pump. Attach a 6-mm outside diameter (OD) glass tee to the flask inlet with a short piece of Teflon tubing. Select a 6-mm OD borosilicate sampling Probe, enlarged at one end to a 12-mm OD and of sufficient length to reach the centroid of the duct to be sampled. Insert a glass wool plug in the enlarged end of the Probe to remove particulate matter. Attach the other end of the Probe to the tee with a short piece of Teflon tubing. Connect a rubber suction bulb to the third leg of the tee. Place the filter end of the Probe at the centroid of the duct, and purge the Probe with the rubber suction bulb. After the Probe is completely purged and filled with duct gases, open the stopcock to the grab flask until the pressure in the flask reaches duct pressure. Close off the stopcock, and remove the Probe from the duct. Remove the tee from the flask and tape the stopcocks to prevent leaks during shipment. Measure and record the duct temperature and pressure.
16.1.3.1.2 Purged Flask Procedure. Attach one end of the sampling flask to a rubber suction bulb. Attach the other end to a 6-mm OD glass Probe as described in Section 8.3.3.1.1. Place the filter end of the Probe at the centroid of the duct, or at a point no closer to the walls than 1 m, and apply suction with the bulb to completely purge the Probe and flask. After the flask has been purged, close off the stopcock near the suction bulb, and then close off the stopcock near the Probe. Remove the Probe from the duct, and disconnect both the Probe and suction bulb. Tape the stopcocks to prevent leakage during shipment. Measure and record the duct temperature and pressure.
16.1.3.2 Flexible Bag Procedure. tedlar or aluminized Mylar bags can also be used to obtain the presurvey sample. Use new bags, and leak-check them before field use. In addition, check the bag before use for contamination by filling it with nitrogen or air, and analyzing the gas by GC at high sensitivity. Experience indicates that it is desirable to allow the inert gas to remain in the bag about 24 hours or longer to check for desorption of organics from the bag. Follow the leak-check and sample collection procedures given in Section 8.2.1.
16.1.3.3 Determination of Moisture Content. For combustion or water- controlled processes, obtain the moisture content from plant personnel or by measurement during the presurvey. If the source is below 59C, measure the wet bulb and dry bulb temperatures, and calculate the moisture content using a psychrometric chart. At higher temperatures, use Method 4 to determine the moisture content.
16.1.4 Determination of Static Pressure.
Obtain the static pressure from the plant personnel or measurement. If a Type S pitot tube and an inclined manometer are used, take care to align the pitot tube 90 from the direction of the flow. Disconnect one of the tubes to the manometer, and read the static pressure; note whether the reading is positive or negative.
16.1.5 Collection of Presurvey Samples with Adsorption Tube.
Follow Section 8.2.4 for presurvey sampling.
17.0 References.
1. American Society for Testing and Materials. C1 Through C5 Hydrocarbons in the Atmosphere by Gas Chromatography. ASTM D 2820-72, Part 23. Philadelphia, Pa. 23:950-958. 1973.
2. Corazon, V.V. Methodology for Collecting and Analyzing Organic Air Pollutants. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA-600/2-79-042. February 1979.
3. Dravnieks, A., B.K. Krotoszynski, J. Whitfield, A. O'Donnell, and T. Burgwald. Environmental Science and Technology. 5(12):1200-1222. 1971.
4. Eggertsen, F.T., and F.M. Nelsen. Gas Chromatographic Analysis of Engine Exhaust and Atmosphere. Analytical Chemistry. 30(6): 1040-1043. 1958.
5. Feairheller, W.R., P.J. Marn, D.H. Harris, and D.L. Harris. Technical Manual for Process Sampling Strategies for Organic Materials. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA 600/2-76-122. April 1976. 172 p.
6. Federal Register, 39 FR 9319-9323. 1974.
7. Federal Register, 39 FR 32857-32860. 1974.
8. Federal Register, 23069-23072 and 23076-23090. 1976.
9. Federal Register, 46569-46571. 1976.
10. Federal Register, 41771-41776. 1977.
11. Fishbein, L. Chromatography of Environmental Hazards, Volume II. Elesevier Scientific Publishing Company. New York, N.Y. 1973.
12. Hamersma, J.W., S.L. Reynolds, and R.F. Maddalone. EPA/IERL-RTP Procedures Manual: Level 1 Environmental Assessment. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA 600/276-160a. June 1976. 130 p.
13. Harris, J.C., M.J. Hayes, P.L. Levins, and D.B. Lindsay. EPA/IERL-RTP Procedures for Level 2 Sampling and Analysis of Organic Materials. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA 600/7-79-033. February 1979. 154 p.
14. Harris, W.E., H.W. Habgood. Programmed temperature Gas Chromatography. John Wiley and Sons, Inc. New York. 1966.
15. Intersociety Committee. Methods of Air Sampling and Analysis. American Health Association. Washington, D.C. 1972.
16. Jones, P.W., R.D. Grammer, P.E. Strup, and T.B. Stanford. Environmental Science and Technology. 10:806-810. 1976.
17. McNair Han Bunelli, E.J. Basic Gas Chromatography. Consolidated Printers. Berkeley. 1969.
18. Nelson, G.O. Controlled Test Atmospheres, Principles and Techniques. Ann Arbor. Ann Arbor Science Publishers. 1971. 247 p.
19. NIOSH Manual of Analytical Methods, Volumes 1, 2, 3, 4, 5, 6, 7. U.S. Department of Health and Human Services, National Institute for Occupational Safety and Health. Center for Disease Control. 4676 Columbia Parkway, Cincinnati, Ohio 45226. April 1977 - August 1981. May be available from the Superintendent of Documents, Government Printing Office, Washington, D.C. 20402. Stock Number/Price:
Volume 1 - O17-033-00267-3/$13
Volume 2 - O17-033-00260-6/$11
Volume 3 - O17-033-00261-4/$14
Volume 4 - O17-033-00317-3/$7.25
Volume 5 - O17-033-00349-1/$10,
Volume 6 - O17-033-00369-6/$9,
Volume 7 - O17-033-00396-5/$7.
Prices subject to change. Foreign orders add 25 percent.
20. Schuetzle, D., T.J. Prater, and S.R. Ruddell. Sampling and Analysis of Emissions from Stationary Sources; I. Odor and Total Hydrocarbons. Journal of the Air Pollution Control Association. 25(9): 925-932. 1975.
21. Snyder, A.D., F.N. Hodgson, M.A. Kemmer and J.R. McKendree. Utility of Solid sorbents for Sampling Organic Emissions from Stationary Sources. U.S. Environmental Protection Agency. Research Triangle Park, N.C. Publication No. EPA 600/2-76-201. July 1976. 71 p.
22. Tentative Method for Continuous Analysis of Total Hydrocarbons in the Atmosphere. Intersociety Committee, American Public Health Association. Washington, D.C. 1972. p. 184-186.
23. Zwerg, G. CRC Handbook of Chromatography, Volumes I and II. Sherma, Joseph (ed.). CRC Press. Cleveland. 1972.
18.0 Tables, Diagrams, flowcharts, and Validation Data.
Figure 18-1. Preliminary survey data sheet.



Figure 18-2. Chromatographic conditions data sheet.

Figure 18-3. Preparation of Standards in Tedlar Bags and calibration Curve.

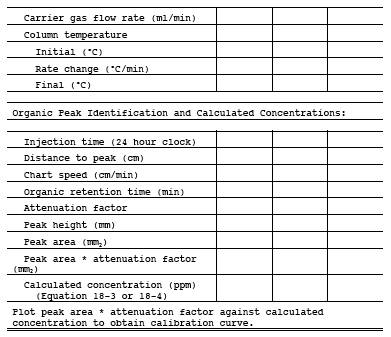
Figure 18-4. flowmeter calibration.


Figure 18-5. Single-Stage calibration Gas Dilution System.
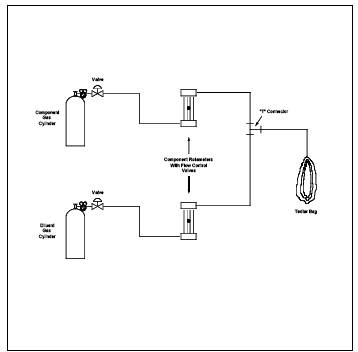
Figure 18-6. Two-Stage Dilution Apparatus.
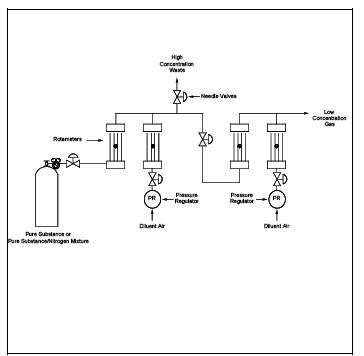
Figure 18-7. Standards prepared by dilution of cylinder standard.
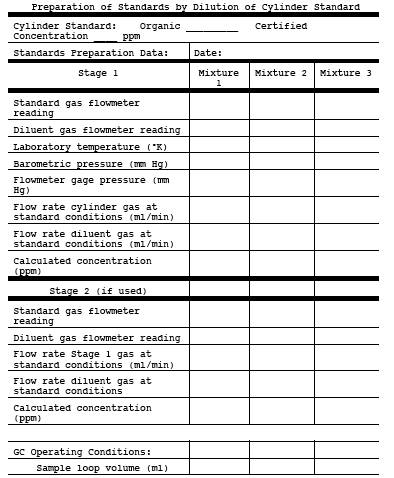
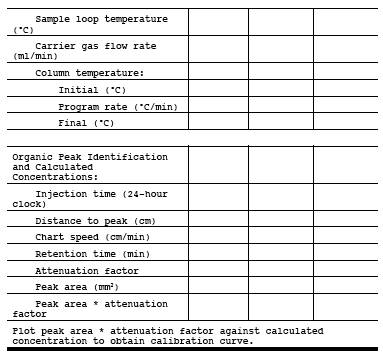
Figure 18-8. Apparatus for Preparation of Liquid Materials.
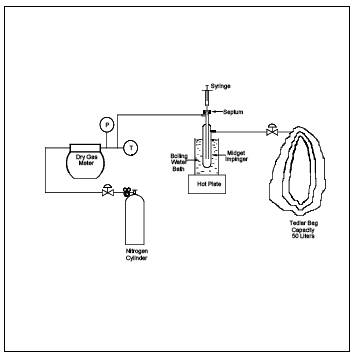
Figure 18-9. Integrated Bag Sampling train.
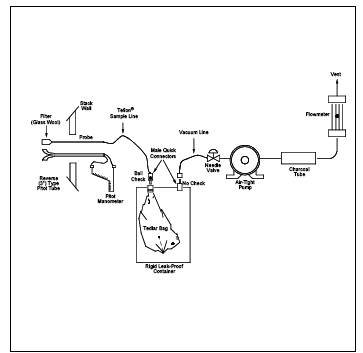
Figure 18-9a. Explosion Risk Gas Sampling Method.
Figure 18-10. Field sample data sheet - Tedlar Bag collection method.

Figure 18-11. Field analysis data sheets.

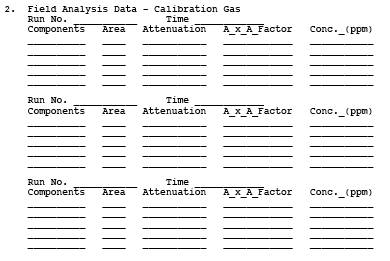
Figure 18-12. Direct Interface Sampling System.
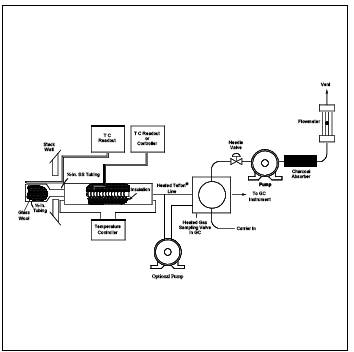
Figure 18-13. Schematic Diagram of the Heated Box Required for Dilution of Sample Gas.
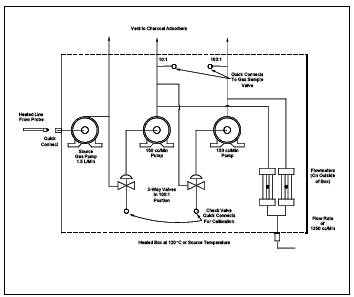
Figure 18-14. Sampling and analysis sheet.

component (3 required)