- Home
- Careers
- Contact
- About
-
Who we are and what we do. -
Press releases, announcements, and notable corporate information. -
We are looking for a few A people. -
We have over 40 years of innovation to create value for our customers. -
We aim to be the highest value provider of every product and service we offer. -
An easy guide to Probe fundamentals.
-
- Services
-
In additional to the analytical results we normally include an expert analysts summary.
We use our decades of experience to help you better understand your data. -
CleanAir can insures that the project goals and testing are objectives are met.
-
We can ship what you need today. It will work. You get a company of experts when you rent from CleanAir. - Thermal Performance
-
- Rental
-
Our factory reconditioning experts work to make old as good as new. -
CleanAir can provide the services required to care of your emissions measurement or power measurement instruments, no matter the age, model or manufacturer.
-
We deliver rental, emergency, or supplemental instruments and onsite services quickly, with minimal operational interruptions .
-
- Products
Featured Product

UL Listed Mobile Temporary Power
Look professional. Don't risk a OSHA fine, or worse causing your customer to get an OSHA or MSHA fine by using an unsafe mobile power distribution system. The CleanAir Temporary Power cart is UL listed! Read more... -
Reference
-
Overviews of products and services -
Learn about our companies and business -
Detailed technical information about the functioning of our products -
Guides and instructions on proper installation and service -
Guides and instructions on proper installation and service -
Drawings, configuration, materials, and limits useful for the planning and layout.
-
CleanAir's reference of video content -
Publications addressing an issue or topic
-
- Site Map
 Express
Express FTIR
FTIR Mercury
Mercury Emission Sampling Equipment
Emission Sampling Equipment Instrument Rental
Instrument RentalEPA Methods List with Links
US EPA Method 101 - Determination Of Particulate And Gaseous Mercury Emissions From Chlor-Alkali Plants (Air Streams)
NOTE: This method does not include all of the specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from methods in Appendix A to 40 CFR Part 60. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 2, Method 3, and Method 5.
Content [ show/hide ].1.0 Scope and Application.
1.1 Analytes.

1.2 Applicability.
This method is applicable for the determination of Hg emissions, including both particulate and gaseous Hg, from chlor-alkali plants and other sources (as specified in the regulations) where the carrier-gas stream in the duct or stack is principally air.
1.3 Data Quality Objectives.
Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method.
Particulate and gaseous Hg emissions are withdrawn isokinetically from the source and collected in acidic iodine monochloride (ICl) solution. The Hg collected (in the mercuric form) is reduced to elemental Hg, which is then aerated from the solution into an optical cell and measured by atomic absorption spectrophotometry.
3.0 Definitions. [Reserved]
4.0 Interferences.
4.1 Sample Collection.
Sulfur dioxide (SO2) reduces ICl and causes premature depletion of the ICl solution.
4.2 Sample Analysis.
4.2.1 ICl concentrations greater than 10-4molar inhibit the reduction of the Hg (II) ion in the aeration cell.
4.2.2 Condensation of water vapor on the optical cell windows causes a positive interference.
5.0 Safety.
5.1 Disclaimer.
This method may involve hazardous materials, operations, and equipment. This test method does not purport to address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive Reagents.
The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burn as thermal burn.
5.2.1 Hydrochloric Acid (HCl).
Highly toxic and corrosive. Causes severe damage to tissues. Vapors are highly irritating to eyes, skin, nose, and lungs, causing severe damage. May cause bronchitis, pneumonia, or edema of lungs. Exposure to concentrations of 0.13 to 0.2 percent can be lethal to humans in a few minutes. Provide ventilation to limit exposure. Reacts with metals, producing hydrogen gas.
5.2.2 Nitric Acid (HNO3).
Highly corrosive to eyes, skin, nose, and lungs. Vapors cause bronchitis, pneumonia, or edema of lungs. Reaction to inhalation may be delayed as long as 30 hours and still be fatal. Provide ventilation to limit exposure. Strong oxidizer. Hazardous reaction may occur with organic materials such as solvents.
5.2.3 Sulfuric Acid (H2SO4).
Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. 3 mg/m3 will cause lung damage. 1 mg/m3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0 Equipment and Supplies.
6.1 Sample Collection.
A schematic of the sampling train used in performing this method is shown inFigure 101-1; it is similar to the Method 5 sampling train. The following items are required for sample collection:
6.1.1 Probe probe nozzle, pitot Tube, Differential Pressure Gauge, metering System, barometer, and Gas Density Determination equipment. Same as Method 5, Sections 6.1.1.1, 6.1.1.3, 6.1.1.4, 6.1.1.9, 6.1.2, and 6.1.3, respectively.
6.1.2 Probe Liner. Borosilicate or quartz glass tubing. A heating system capable of maintaining a gas temperature of 120 ± 14 C (248 ± 25 F) at the Probe exit during sampling may be used to prevent water condensation.
NOTE: Do not use metal Probe liners.
6.1.3 impingers. Four Greenburg-Smith impingers connected in series with leak-free ground glass fittings or any similar leak-free non-contaminating fittings. For the
first, third, and fourth impingers, impingers that are modified by replacing the tip with a 13-mm ID (0.5-in.) glass tube extending to 13 mm (0.5 in.) from the bottom of the flask may be used.
6.1.4 Acid Trap. Mine Safety Appliances air line filter, Catalog number 81857, with acid absorbing cartridge and suitable connections, or equivalent.
6.2 Sample Recovery.
The following items are needed for sample recovery:
6.2.1 glass Sample Bottles. Leakless, with Teflon-lined caps, 1000- and 100-ml.
6.2.2 Graduated Cylinder. 250-ml.
6.2.3 Funnel and Rubber Policeman. To aid in transfer of silica gel to container; not necessary if silica gel is weighed in the field.
6.2.4 Funnel. glass, to aid in sample recovery.
6.3 Sample Preparation and Analysis.
The following items are needed for sample preparation and analysis:
6.3.1 Atomic Absorption Spectrophotometer. Perkin-Elmer 303, or equivalent, containing a hollow-cathode mercury lamp and the optical cell described in Section 6.3.2.
6.3.2 Optical Cell. Cylindrical shape with quartz end windows and having the dimensions shown in Figure 101-2. Wind the cell with approximately 2 meters (6 ft) of 24-gauge Nichrome wire, or equivalent, and wrap with fiberglass insulation tape, or equivalent; do not let the wires touch each other.
6.3.3 Aeration Cell. Constructed according to the specifications in Figure 101-3. Do not use a glass frit as a substitute for the blown glass bubbler tip shown in Figure 101-3.
6.3.4 Recorder. Matched to output of the spectrophotometer described in Section 6.3.1.
6.3.5 Variable Transformer. To vary the voltage on the optical cell from 0 to 40 volts.
6.3.6 Hood. For venting optical cell exhaust.
6.3.7 flow metering Valve.
6.3.8 Rate meter. Rotameter, or equivalent, capable of measuring to within 2 percent a gas flow of 1.5 liters/min (0.053 cfm).
6.3.9 Aeration Gas Cylinder. Nitrogen or dry, Hg-free air, equipped with a single-stage regulator.
6.3.10 tubing. For making connections. Use glass tubing (ungreased ball and socket connections are recommended) for all tubing connections between the solution cell and the optical cell; do not use Tygon tubing, other types of flexible tubing, or metal tubing as substitutes. Teflon, steel, or copper tubing may be used between the nitrogen tank and flow metering valve (Section 6.3.7), and Tygon, gum, or rubber tubing between the flow metering valve and the aeration cell.
6.3.11 flow Rate calibration equipment. Bubble flow meter or wet-test meter for measuring a gas flow rate of 1.5 ± 0.1 liters/min (0.053 ± 0.0035 cfm).
6.3.12 Volumetric Flasks. Class A with penny head standard taper stoppers; 100-, 250-, 500-, and 1000-ml.
6.3.13 Volumetric Pipets. Class A; 1-, 2-, 3-, 4-, and 5-ml.
6.3.14 Graduated Cylinder. 50-ml.
6.3.15 Magnetic Stirrer. General-purpose laboratory type.
6.3.16 Magnetic Stirring Bar. Teflon-coated.
6.3.17 Balance. Capable of weighing to ± 0.5 g
6.3.18 Alternative Analytical Apparatus. Alternative systems are allowable as long as they meet the following criteria:
6.3.18.1 A linear calibration curve is generated and two consecutive samples of the same aliquot size and concentration agree within 3 percent of their average.
6.3.18.2 A minimum of 95 percent of the spike is recovered when an aliquot of a source sample is spiked with a known concentration of Hg (II) compound.
6.3.18.3 The reducing agent should be added after the aeration cell is closed.
6.3.18.4 The aeration bottle bubbler should not contain a frit.
6.3.18.5 Any Tygon tubing used should be as short as possible and conditioned prior to use until blanks and standards yield linear and reproducible results.
6.3.18.6 If manual stirring is done before aeration, it should be done with the aeration cell closed.
6.3.18.7 A drying tube should not be used unless it is conditioned as the Tygon tubing above.
7.0 Reagents and Standards.
Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society; where such specifications are not available, use the best available grade.
7.1 Sample Collection.
The following reagents are required for sample collection:
7.1.1 Water. Deionized distilled, to conform to ASTM D 1193-77 or 91 (incorporated by reference - see 61.18), Type 1. If high concentrations of organic matter are not expected to be present, the analyst may eliminate the KMnO4 test for oxidizable organic matter. Use this water in all dilutions and solution preparations.
7.1.2 Nitric Acid, 50 Percent (v/v). Mix equal volumes of concentrated HNO3 and water, being careful to add the acid to the water slowly.
7.1.3 Silica Gel. Indicating type, 6- to 16-mesh. If previously used, dry at 175 C (350 F) for 2 hours. The tester may use new silica gel as received.
7.1.4 Potassium Iodide (KI) Solution, 25 Percent. Dissolve 250 g of KI in water, and dilute to 1 liter.
7.1.5 Iodine Monochloride Stock Solution, 1.0 M. To 800 ml of 25 percent KI solution, add 800 ml of concentrated HCl. Cool to room temperature. With vigorous stirring, slowly add 135 g of potassium iodate (KIO3), and stir until all free iodine has dissolved. A clear orange-red solution occurs when all the KIO3 has been added. Cool to room temperature, and dilute to 1800 ml with water. Keep the solution in amber glass bottles to prevent degradation.
7.1.6 Absorbing Solution, 0.1 M ICl. Dilute 100 ml of the 1.0 M ICl stock solution to 1 liter with water. Keep the solution in amber glass bottles and in darkness to prevent degradation. This reagent is stable for at least two months.
7.2 Sample Preparation and Analysis.
The following reagents and standards are required for sample preparation and analysis:
7.2.1 Reagents
7.2.1.1 Tin (II) Solution. Prepare fresh daily, and keep sealed when not being used. Completely dissolve 20 g of tin (II) chloride [or 25 g of tin (II) sulfate] crystals (Baker Analyzed reagent grade or any other brand that will give a clear solution) in 25 ml of concentrated HCl. Dilute to 250 ml with water. Do not substitute HNO3, H2SO>4, or other strong acids for the HCl.
7.2.1.2 Sulfuric Acid, 5 Percent (v/v). Dilute 25 ml of concentrated H2SO4 to 500 ml with water.
7.2.2 Standards
7.2.2.1 Hg Stock Solution, 1 mg Hg/ml. Prepare and store all Hg standard solutions in borosilicate glass containers. Completely dissolve 0.1354 g of Hg (II) chloride in 75 ml of water in a 100-ml glass volumetric flask. Add 10 ml of concentrated HNO3, and adjust the volume to exactly 100 ml with water. Mix thoroughly. This solution is stable for at least one month.
7.2.2.2 Intermediate Hg Standard Solution, 10 g Hg/ml. Prepare fresh weekly. Pipet 5.0 ml of the Hg stock solution (Section 7.2.2.1) into a 500-ml glass volumetric flask, and add 20 ml of the 5 percent H2SO4 solution. Dilute to exactly 500 ml with water. Thoroughly mix the solution.
7.2.2.3 Working Hg Standard Solution, 200 ng Hg/ml. Prepare fresh daily. Pipet 5.0 ml of the intermediate Hg standard solution (Section 7.2.2.2) into a 250-ml volumetric glass flask. Add 10 ml of the 5 percent H2SO>4 and 2 ml of the 0.1 M ICl absorbing solution taken as a blank (Section 8.7.4.3), and dilute to 250 ml with water. Mix thoroughly.
8.0 Sample Collection, Preservation, Transport, and Storage.
Because of the complexity of this method, testers should be trained and experienced with the test procedures to ensure reliable results. Since the amount of Hg that is collected generally is small, the method must be carefully applied to prevent contamination or loss of sample.
8.1 Pretest Preparation.
Follow the general procedure outlined in Method 5, Section 8.1, except omit Sections 8.1.2 and 8.1.3.
8.2 Preliminary Determinations.
Follow the general procedure outlined in Method 5, Section 8.2, with the exception of the following:
8.2.1 Select a probe nozzle size based on the range of velocity heads to assure that it is not necessary to change the probe nozzle size in order to maintain isokinetic sampling rates below 28 liters/min (1.0 cfm).
8.2.2 Perform test runs such that samples are obtained over a period or periods that accurately determine the maximum emissions that occur in a 24-hour period. In the case of cyclic operations, run sufficient tests for the accurate determination of the emissions that occur over the duration of the cycle. A minimum sample time of 2 hours is recommended. In some instances, high Hg or high SO2 concentrations make it impossible to sample for the desired minimum time. This is indicated by reddening (liberation of free iodine) in the first impinger. In these cases, the sample run may be divided into two or more sub-runs to ensure that the absorbing solution is not depleted.
8.3 Preparation of Sampling Train.
8.3.1 Clean all glassware (Probe, impingers, and connectors) by rinsing with 50 percent HNO3, tap water, 0.1 M ICl, tap water, and finally deionized distilled water. Place 100 ml of 0.1 M ICl in each of the first three impingers. Take care to prevent the absorbing solution from contacting any greased surfaces. Place approximately 200 g of pre-weighed silica gel in the fourth impinger. More silica gel may be used, but care should be taken to ensure that it is not entrained and carried out from the impinger during sampling. Place the silica gel container in a clean place for later use in the sample recovery. Alternatively, determine and record the weight of the silica gel plus impinger to the nearest 0.5 g.
8.3.2 Install the selected probe nozzle using a Viton A O-ring when stack temperatures are less than 260 C (500 F). Use a fiberglass string gasket if temperatures are higher. See APTD-0576 (Reference 3 in Method 5) for details. Other connecting systems using either 316 stainless steel or Teflon ferrules may be used. Mark the Probe with heat-resistant tape or by some other method to denote the proper distance into the stack or duct for each sampling point.
8.3.3 Assemble the train as shown in Figure 101-1, using (if necessary) a very light coat of silicone grease on all ground glass joints. Grease only the outer portion (see APTD-0576) to avoid the possibility of contamination by the silicone grease.
NOTE: An empty impinger may be inserted between the third impinger and the silica gel to remove excess moisture from the sample stream.
8.3.4 After the sampling train has been assembled, turn on and set the Probe heating system, if applicable, at the desired operating temperature. Allow time for the temperatures to stabilize. Place crushed ice around the impingers.
8.4 Leak-Check Procedures.
Follow the leak-check procedures outlined in Method 5, Section 8.4.
8.5 Sampling Train Operation.
Follow the general procedure outlined in Method 5, Section 8.5. For each run, record the data required on a data sheet such as the one shown in Figure 101-4.
8.6 Calculation of Percent Isokinetic.
Same as Method 5, Section 8.6.
8.7 Sample Recovery.
Begin proper cleanup procedure as soon as the Probe is removed from the stack at the end of the sampling period.
8.7.1 Allow the Probe to cool. When it can be safely handled, wipe off any external particulate matter near the tip of the Probe probe nozzle, and place a cap over it. Do not cap off the Probe tip tightly while the sampling train is cooling. Capping would create a vacuum and draw liquid out from the impingers.
8.7.2 Before moving the sampling train to the cleanup site, remove the Probe from the train, wipe off the silicone grease, and cap the open outlet of the Probe. Be careful not to lose any condensate that might be present. Wipe off the silicone grease from the impinger. Use either ground-glass stoppers, plastic caps, or serum caps to close these openings.
8.7.3 Transfer the Probe and impinger assembly to a cleanup area that is clean, protected from the wind, and free of Hg contamination. The ambient air in laboratories located in the immediate vicinity of Hg-using facilities is not normally free of Hg contamination.
8.7.4 Inspect the train before and during disassembly, and note any abnormal conditions. Treat the samples as follows.
8.7.4.1 Container No. 1 (impingers and Probe).
8.7.4.1.1 Using a graduated cylinder, measure the liquid in the first three impingers to within 1 ml. Record the volume of liquid present (e.g., see Figure 5-6 of Method 5). This information is needed to calculate the moisture content of the effluent gas. (Use only glass storage bottles and graduated cylinders that have been precleaned as in Section 8.3.1) Place the contents of the first three impingers into a 1000-ml glass sample bottle.
8.7.4.1.2 Taking care that dust on the outside of the Probe or other exterior surfaces does not get into the sample, quantitatively recover the Hg (and any condensate) from the Probe probe nozzle, Probe fitting, and Probe liner as follows: Rinse these components with two 50-ml portions of 0.1 M ICl. Next, rinse the Probe probe nozzle, fitting and liner, and each piece of connecting glassware between the Probe liner and the back half of the third impinger with a maximum of 400 ml of water. Add all washings to the 1000-ml glass sample bottle containing the liquid from the first three impingers.
8.7.4.1.3 After all washings have been collected in the sample container, tighten the lid on the container to prevent leakage during shipment to the laboratory. Mark the height of the liquid to determine later whether leakage occurred during transport. Label the container to identify clearly its contents.
8.7.4.2 Container No. 2 (Silica Gel). Same as Method 5, Section 8.7.6.3.
8.7.4.3 Container No. 3 (Absorbing Solution Blank). Place 50 ml of the 0.1 M ICl absorbing solution in a 100-ml sample bottle. Seal the container. Use this blank to prepare the working Hg standard solution (Section 7.2.2.3).
9.0 Quality Control.
9.1 Miscellaneous Quality Control Measures.
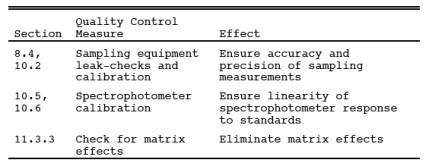
9.2 Volume metering System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and Standardizations.
NOTE: Maintain a laboratory log of all calibrations.
10.1 Before Use
Before use, clean all glassware, both new and used, as follows: brush with soap and tap water, liberally rinse with tap water, soak for 1 hour in 50 percent HNO3, and then rinse with deionized distilled water.
10.2 Sampling equipment.
Calibrate the sampling equipment according to the procedures outlined in the following sections of Method 5: Section 10.1 (Probe probe nozzle), Section 10.2 (pitot Tube Assembly), Section 10.3 (metering System), Section 10.5 (tenperature sensors), Section 10.6 (barometer).
10.3 Aeration System flow Rate Meter.
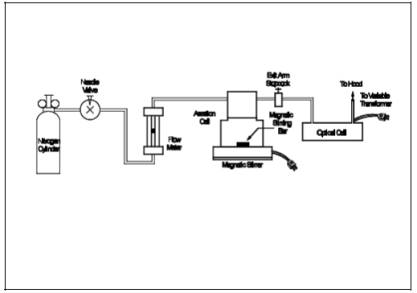
Assemble the aeration system as shown in Figure 101-5. Set the outlet pressure on the aeration gas cylinder regulator to a minimum pressure of 500 mm Hg (10 psi), and use the flowmetering valve and a bubble flowmeter or wet-test meter to obtain a flow rate of 1.5 ± 0.1 liters/min (0.053 ± 0.0035 cfm) through the aeration cell. After the calibration of the aeration system flow rate meter is complete, remove the bubble flowmeter from the system.
10.4 Optical Cell Heating System.
Using a 50-ml graduated cylinder, add 50 ml of water to the bottle section of the aeration cell, and attach the bottle section to the bubbler section of the cell. Attach the aeration cell to the optical cell and while aerating at 1.5 ± 0.1 liters/min (0.053 ± 0.0035 cfm), determine the minimum variable transformer setting necessary to prevent condensation of moisture in the optical cell and in the connecting tubing. (This setting should not exceed 20 volts.)
10.5 Spectrophotometer and Recorder.
10.5.1 The Hg response may be measured by either peak height or peak area.
NOTE: The temperature of the solution affects the rate at which elemental Hg is released from a solution and, consequently, it affects the shape of the absorption curve (area) and the point of maximum absorbance (peak height). Therefore, to obtain reproducible results, bring all solutions to room temperature before use.
10.5.2 Set the spectrophotometer wavelength at 253.7 nm, and make certain the optical cell is at the minimum temperature that will prevent water condensation. Then set the recorder scale as follows: Using a 50-ml graduated cylinder, add 50 ml of water to the aeration cell bottle. Add three drops of Antifoam B to the bottle, and then pipet 5.0 ml of the working Hg standard solution into the aeration cell.
NOTE: Always add the Hg-containing solution to the aeration cell after the 50 ml of water.
10.5.3 Place a Teflon-coated stirring bar in the bottle. Before attaching the bottle section to the bubbler section of the aeration cell, make certain that (1) the aeration cell exit arm stopcock (Figure 101-3) is closed (so that Hg will not prematurely enter the optical cell when the reducing agent is being added) and (2) there is no flow through the bubbler. If conditions (1) and (2) are met, attach the bottle section to the bubbler section of the aeration cell. Pipet 5 ml of tin (II) reducing solution into the aeration cell through the side arm, and immediately stopper the side arm. Stir the solution for 15 seconds, turn on the recorder, open the aeration cell exit arm stopcock, and immediately initiate aeration with continued stirring. Determine the maximum absorbance of the standard, and set this value to read 90 percent of the recorder full scale.
10.6 calibration Curve.
10.6.1 After setting the recorder scale, repeat the procedure in Section 10.5 using 0.0-, 1.0-, 2.0-, 3.0-, 4.0-, and 5.0-ml aliquots of the working standard solution (final amount of Hg in the aeration cell is 0, 200, 400, 600, 800, and 1000 ng, respectively). Repeat this procedure on each aliquot size until two consecutive peaks agree within 3 percent of their average value.
NOTE: To prevent Hg carryover from one sample to another, do not close the aeration cell from the optical cell until the recorder pen has returned to the baseline.) 10.6.2 It should not be necessary to disconnect the aeration gas inlet line from the aeration cell when changing samples. After separating the bottle and bubbler sections of the aeration cell, place the bubbler section into a 600-ml beaker containing approximately 400 ml of water. Rinse the bottle section of the aeration cell with a stream of water to remove all traces of the tin (II) reducing agent. Also, to prevent the loss of Hg before aeration, remove all traces of the reducing agent between samples by washing with water. It will be necessary, however, to wash the aeration cell parts with concentrated HCl if any of the following conditions occur: (1) A white film appears on any inside surface of the aeration cell, (2) the calibration curve changes suddenly, or (3) the replicate samples do not yield reproducible results.
10.6.3 Subtract the average peak height (or peak area) of the blank (0.0-ml aliquot) - which must be less than 2 percent of recorder full scale - from the averaged peak heights of the 1.0-, 2.0-, 3.0-, 4.0-, and 5.0-ml aliquot standards. If the blank absorbance is greater than 2 percent of full-scale, the probable cause is Hg contamination of a reagent or carry-over of Hg from a previous sample. Prepare the calibration curve by plotting the corrected peak height of each standard solution versus the corresponding final total Hg weight in the aeration cell (in ng), and draw the best fit straight line. This line should either pass through the origin or pass through a point no further from the origin than ± 2 percent of the recorder full scale. If the line does not pass through or very near to the origin, check for nonlinearity of the curve and for incorrectly prepared standards.
11.0 Analytical Procedure.
11.1 Sample Loss Check.
Check the liquid level in each container to see whether liquid was lost during transport. If a noticeable amount of leakage occurred, either void the sample or use methods subject to the approval of the Administrator to account for the losses.
11.2 Sample Preparation.
Treat each sample as follows:
11.2.1 Container No. 1 (impingers and Probe). Carefully transfer the contents of Container No. 1 into a 1000-ml volumetric flask, and adjust the volume to exactly 1000 ml with water.
11.2.2 Dilutions. Pipet a 2-ml aliquot from the diluted sample from Section 11.2.1 into a 250-ml volumetric flask. Add 10 ml of 5 percent H2SO>4, and adjust the volume to exactly 250 ml with water. This solution is stable for at least 72 hours.
NOTE: The dilution factor will be 250/2 for this solution.
11.3 Analysis.
Calibrate the analytical equipment and develop a calibration curve as outlined in Sections 10.3 through 10.6.
11.3.1 Mercury Samples. Repeat the procedure used to establish the calibration curve with an appropriately sized aliquot (1 to 5 ml) of the diluted sample (from Section 11.2.2) until two consecutive peak heights agree within 3 percent of their average value. The peak maximum of an aliquot (except the 5-ml aliquot) must be greater than 10 percent of the recorder full scale. If the peak maximum of a 1.0-ml aliquot is off scale on the recorder, further dilute the original source sample to bring the Hg concentration into the calibration range of the spectrophotometer.
11.3.2 Run a blank and standard at least after every five samples to check the spectrophotometer calibration. The peak height of the blank must pass through a point no further from the origin than ±2 percent of the recorder full scale. The difference between the measured concentration of the standard (the product of the corrected peak height and the reciprocal of the least squares slope) and the actual concentration of the standard must be less than 7 percent, or recalibration of the analyzer is required.
11.3.3 Check for Matrix Effects (optional). Use the Method of Standard Additions as follows to check at least one sample from each source for matrix effects on the Hg results. The Method of Standard Additions procedures described on pages 9-4 and 9-5 of the section entitled "General Information" of the Perkin Elmer Corporation Atomic Absorption Spectrophotometry Manual, Number 303-0152 (Reference 16 in Section 16.0) are recommended. If the results of the Method of Standard Additions procedure used on the single source sample do not agree to within ±5 percent of the value obtained by the routine atomic absorption analysis, then reanalyze all samples from the source using the Method of Standard Additions procedure.
11.4 Container No. 2 (Silica Gel).
Weigh the spent silica gel (or silica gel plus impinger) to the nearest 0.5 g using a balance. (This step may be conducted in the field.)
12.0 Data Analysis and Calculations.
Carry out calculations, retaining at least one extra decimal significant figure beyond that of the acquired data. Round off figures only after the final calculation. Other forms of the equations may be used as long as they give equivalent results.
12.1 Average console meter temperature and Average Orifice Pressure Drop, Dry Gas Volume, Volume of Water Vapor Condensed, Moisture Content, and Isokinetic Variation. Same as Method 5, Sections 12.2 through 12.5 and 12.11, respectively.
12.2 Stack Gas Velocity. Using the data from this test and Equation 2-9 of Method 2, calculate the average stack gas velocity vs.
12.3.1 For each source sample, correct the average maximum absorbance of the two consecutive samples whose peak heights agree within 3 percent of their average for the contribution of the solution blank (see Section 10.6.3). Use the calibration curve and these corrected averages to determine the final total weight of Hg in ng in the aeration cell for each source sample.
12.3.2 Correct for any dilutions made to bring the sample into the working range of the spectrophotometer. Then calculate the Hg in the original solution, mHg, as follows:
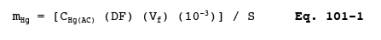
where:

12.4 Mercury Emission Rate. Calculate the daily Hg emission rate, R, using Equation 101-2. For continuous operations, the operating time is equal to 86,400 seconds per day. For cyclic operations, use only the time per day each stack is in operation. The total Hg emission rate from a source will be the summation of results from all stacks.
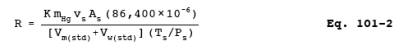
where:

12.5 Determination of Compliance. Each performance test consists of three repetitions of the applicable test method. For the purpose of determining compliance with an applicable national emission standard, use the average of the results of all repetitions.
13.0 Method Performance.
The following estimates are based on collaborative tests, wherein 13 laboratories performed duplicate analyses on two Hg-containing samples from a chlor-alkali plant and on one laboratory-prepared sample of known Hg concentration. The sample concentrations ranged from 2 to 65 g Hg/ml.
13.1 Precision. The estimated intra-laboratory and inter-laboratory standard deviations are 1.6 and 1.8 g Hg/ml, respectively.
13.2 Accuracy. The participating laboratories that analyzed a 64.3 g Hg/ml (in 0.1 M ICl) standard obtained a mean of 63.7 g Hg/ml.
13.3 Analytical Range. After initial dilution, the range of this method is 0.5 to 120 g Hg/ml. The upper limit can be extended by further dilution of the sample.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
Same as Method 5, Section 17.0, References 1-3, 5, and 6, with the addition of the following:
1. Determining Dust Concentration in a Gas Stream. ASME Performance Test Code No. 27. New York, NY. 1957.
2. DeVorkin, Howard, et al. Air Pollution Source Testing Manual. Air Pollution Control District. Los Angeles, CA. November 1963.
3. Hatch, W.R., and W.I. Ott. Determination of Sub-Microgram Quantities of Mercury by Atomic Absorption Spectrophotometry. Anal. Chem. 40:2085-87. 1968.
4. Mark, L.S. Mechanical Engineers' Handbook. McGraw-Hill Book Co., Inc. New York, NY. 1951.
5. Western Precipitation Division of Joy Manufacturing Co. Methods for Determination of Velocity, Volume, Dust and Mist Content of Gases. Bulletin WP-50. Los Angeles, CA. 1968.
6. Perry, J.H. Chemical Engineers' Handbook. McGraw-Hill Book Co., Inc. New York, NY. 1960.
7. Shigehara, R.T., W.F. Todd, and W.S. Smith. Significance of Errors in Stack Sampling Measurements. Stack Sampling News. 1(3):6-18. September 1973.
8. Smith, W.S., R.T. Shigehara, and W.F. Todd. A Method of Interpreting Stack Sampling Data. Stack Sampling News. 1(2):8-17. August 1973.
9. Standard Method for Sampling Stacks for Particulate Matter. In: 1971 Annual Book of ASTM Standards, Part 23. ASTM Designation D 2928-71. Philadelphia, PA 1971.
10. Vennard, J.K. Elementary Fluid Mechanics. John Wiley and Sons, Inc. New York. 1947.
11. Mitchell, W.J. and M.R. Midgett. Improved Procedure for Determining Mercury Emissions from Mercury Cell Chlor-Alkali Plants. J. APCA. 26:674-677. July 1976.
12. Shigehara, R.T. Adjustments in the EPA Nomograph for Different pitot Tube Coefficients and Dry Molecular Weights. Stack Sampling News. 2:4-11. October 1974.
13. Vollaro, R.F. Recommended Procedure for Sample Traverses in Ducts Smaller than 12 Inches in Diameter. U.S. Environmental Protection Agency, Emission Measurement Branch. Research Triangle Park, NC. November 1976.
14. Klein, R. and C. Hach. Standard Additions: Uses and Limitation in Spectrophotometric Measurements. Amer. Lab. 9:21. 1977.
15. Perkin Elmer Corporation. Analytical Methods for Atomic Absorption Spectrophotometry. Norwalk, Connecticut. September 1976.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
Figure 101-1. Mercury Sampling Train.
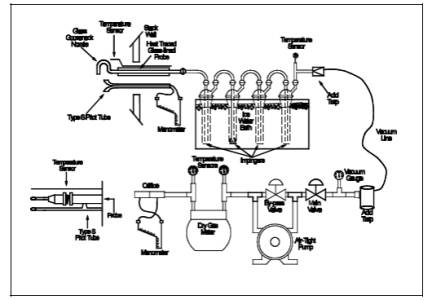
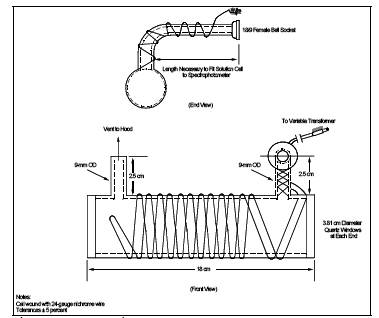
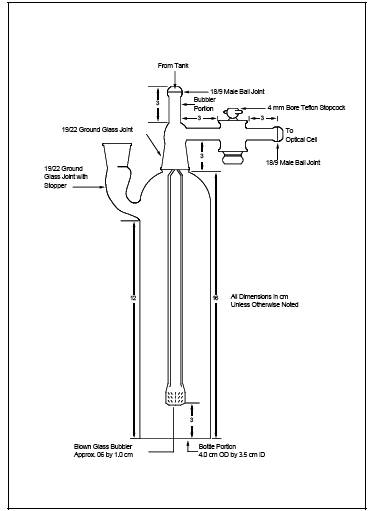
Figure 101-4. Mercury Field Data.
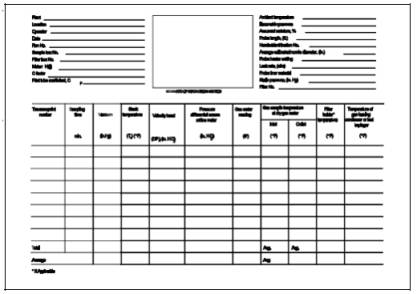
Figure 101-5. Schematic of Aeration System.
- Analytical
- Ion Chromatography
- Gas Chromatography
- Gravimetrics
- Ash Resistivity
- Inks/Coatings
- Scrubber Stoichiometry
- Titrations
- Mercury Sorbent Trap
- Engineering
- Express Products
- Rental Instruments
- MET80 Mercury Monitor
- Continuous Emission Monitors
- Gas Sampling Equipment
- Mobile Power Supply
Our Resources
- Technical Resources