Warning: include(common/header.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 22
Warning: include(): Failed opening 'common/header.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 22
Warning: include(common-home/leftcolumn_epa.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 30
Warning: include(): Failed opening 'common-home/leftcolumn_epa.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 30
EPA Methods List with Links
US EPA Method 26A - Determination Of Hydrogen Halide And Halogen Emissions From Stationary Sources Isokinetic Method
NOTE: This method does not include all of the specifications (e.g. equipment and supplies) and procedures (e.g. sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 2, Method 5, and Method 26.
Content [ show/hide ].
1.0 Scope and Application.
1.1 Analytes.
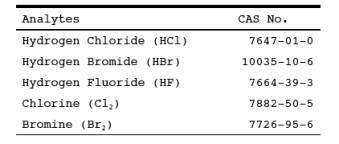
1.2 This method is applicable for determining emissions of hydrogen halides (HX) [HCl, HBr, and HF] and halogens (X2) [Cl2 and Br>2] from stationary sources when specified by the applicable subpart. This method collects the emission sample isokinetically and is therefore particularly suited for sampling at sources, such as those controlled by wet scrubbers, emitting acid particulate matter (e.g., hydrogen halides dissolved in water droplets).
1.3 Data Quality Objectives.
Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method.
2.1 Principle.
Gaseous and particulate pollutants are withdrawn isokinetically from the source and collected in an optional cyclone, on a filter, and in absorbing solutions. The cyclone collects any liquid droplets and is not necessary if the source emissions do not contain them; however, it is preferable to include the cyclone in the sampling train to protect the filter from any liquid present. The filter collects particulate matter including halide salts but is not routinely recovered or analyzed. Acidic and alkaline absorbing solutions collect the gaseous hydrogen halides and halogens, respectively. Following sampling of emissions containing liquid droplets, any halides/halogens dissolved in the liquid in the cyclone and on the filter are vaporized to gas and collected in the impingers by pulling conditioned ambient air through the sampling train. The hydrogen halides are solubilized in the acidic solution and form chloride (Cl-), bromide (Br-), and fluoride (F-) ions. The halogens have a very low solubility in the acidic solution and pass through to the alkaline solution where they are hydrolyzed to form a proton (H+), the halide ion, and the hypohalous acid (HClO or HBrO). Sodium thiosulfate is added to the alkaline solution to assure reaction with the hypohalous acid to form a second halide ion such that 2 halide ions are formed for each molecule of halogen gas. The halide ions in the separate solutions are measured by ion chromatography (IC). If desired, the particulate matter recovered from the filter and the Probe is analyzed following the procedures in Method 5.
NOTE: If the tester intends to use this sampling arrangement to sample concurrently for particulate matter, the alternative Teflon Probe liner, cyclone, and filter holder should not be used. The Teflon filter support must be used. The tester must also meet the Probe and filter temperature requirements of both sampling trains.
3.0 Definitions. [Reserved]
4.0 Interferences.
4.1 Volatile materials, such as chlorine dioxide (ClO2) and ammonium chloride (NH4Cl), which produce halide ions upon dissolution during sampling are potential interferents. Interferents for the halide measurements are the halogen gases which disproportionate to a hydrogen halide and a hypohalous acid upon dissolution in water. The use of acidic rather than neutral or basic solutions for collection of the hydrogen halides greatly reduces the dissolution of any halogens passing through this solution.
4.2 The simultaneous presence of both HBr and Cl2 may cause a positive bias in the HCl result with a corresponding negative bias in the Cl2 result as well as affecting the HBr/Br2split.
4.3 High concentrations of nitrogen oxides (NOx) may produce sufficient nitrate (NO3-) to interfere with measurements of very low Br>- levels.
4.4 There is anecdotal evidence that HF may be outgassed from new Teflon components. If HF is a target analyte then preconditioning of new Teflon components, by heating, should be considered.
5.0 Safety.
5.1 Disclaimer.
This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicability of regulatory limitations before performing this test method.
5.2 Corrosive Reagents.
The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Sodium Hydroxide (NaOH). Causes severe damage to eyes and skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.2 Sulfuric Acid (H2SO4). Rapidly destructive to body tissue. Will cause third degree burns. Eye damage may result in blindness. Inhalation may be fatal from spasm of the larynx, usually within 30 minutes. May cause lung tissue damage with edema. 1 mg/m>3 for 8 hours will cause lung damage or, in higher concentrations, death. Provide ventilation to limit inhalation. Reacts violently with metals and organics.
6.0. equipment and supplies.
NOTE: Mention of trade names or specific products does not constitute endorsement by the Environmental Protection Agency.
6.1 Sampling.
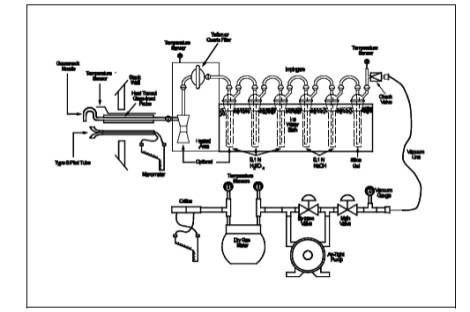
The sampling train is shown in Figure 26A-1; the apparatus is similar to the Method 5 train where noted as follows:
6.1.1 Probe probe nozzle.
Borosilicate or quartz glass; constructed and calibrated according to Method 5, Sections 6.1.1.1and 10.1, and coupled to the Probe liner using a Teflon union; a stainless steel nut is recommended for this union. When the stack temperature exceeds 210C (410F), a one-piece glass probe nozzle/liner assembly must be used.
6.1.2 Probe Liner.
Same as Method 5, Section 6.1.1.2, except metal liners shall not be used. Watercooling of the stainless steel sheath is recommended at temperatures exceeding 500C (932F). Teflon may be used in limited applications where the minimum stack temperature exceeds 120C (250F) but never exceeds the temperature where Teflon is estimated to become unstable [approximately 210C (410F)].
6.1.3 pitot Tube, Differential Pressure Gauge, filter Heating System, metering System, barometer, Gas Density Determination equipment.
Same as Method 5, Sections 6.1.1.3, 6.1.1.4, 6.1.1.6, 6.1.1.9, 6.1.2, and 6.1.3.
6.1.4 Cyclone (Optional). glass or Teflon.
Use of the cyclone is required only when the sample gas stream is saturated with moisture; however, the cyclone is recommended to protect the filter from any liquid droplets present.
6.1.5 filter Holder.
Borosilicate or quartz glass, or Teflon filter holder, with a Teflon filter support and a sealing gasket. The sealing gasket shall be constructed of Teflon or equivalent materials. The holder design shall provide a positive seal against leakage at any point along the filter circumference. The holder shall be attached immediately to the outlet of the cyclone.
6.1.6 impinger train.
The following system shall be used to determine the stack gas moisture content and to collect the hydrogen halides and halogens: five or six impingers connected in series with leak-free ground glass fittings or any similar leak-free noncontaminating fittings. The first impinger shown in Figure 26A-1 (knockout or condensate impinger) is optional and is recommended as a water knockout trap for use under high moisture conditions. If used, this impinger should be constructed as described below for the alkaline impingers, but with a shortened stem, and should contain 50 ml of 0.1 N H2SO4. The following two impingers (acid impingers which each contain 100 ml of 0.1 N H2SO>4) shall be of the Greenburg-Smith design with the standard tip (Method 5, Section 6.1.1.8). The next two impingers (alkaline impingers which each contain 100 ml of 0.1 N NaOH) and the last impinger (containing silica gel) shall be of the modified Greenburg-Smith design (Method 5, Section 6.1.1.8). The condensate, acid, and alkaline impingers shall contain known quantities of the appropriate absorbing reagents. The last impinger shall contain a known weight of silica gel or equivalent desiccant. Teflon impingers are an acceptable alternative.
6.1.7 Heating System.
Any heating system capable of maintaining a temperature around the Probe and filter holder greater than 120 C (248 F) during sampling, or such other temperature as specified by an applicable subpart of the standards or approved by the Administrator for a particular application.
6.1.8 Ambient Air Conditioning Tube (Optional).
Tube tightly packed with approximately 150 g of fresh 8 to 20 mesh sodium hydroxide-coated silica, or equivalent, (Ascarite II has been found suitable) to dry and remove acid gases from the ambient air used to remove moisture from the filter and cyclone, when the cyclone is used. The inlet and outlet ends of the tube should be packed with at least 1-cm thickness of glass wool or filter material suitable to prevent escape of fines. Fit one end with flexible tubing, etc. to allow connection to Probe probe nozzle following the test run.
6.2 Sample Recovery.
6.2.1 Probe-Liner and Probe-probe nozzle Brushes, Wash Bottles, glass Sample Storage Containers, Petri Dishes, Graduated Cylinder and/or Balance, and Rubber Policeman. Same as Method 5, Sections 6.2.1, 6.2.2, 6.2.3, 6.2.4, 6.2.5, and 6.2.7.
6.2.2 Plastic Storage Containers. Screw-cap polypropylene or polyethylene containers to store silica gel. High-density polyethylene bottles with Teflon screw cap liners to store impinger reagents, 1-liter.
6.2.3 Funnels. glass or high-density polyethylene, to aid in sample recovery.
6.3 Sample Preparation and Analysis.
6.3.1 Volumetric Flasks. Class A, various sizes.
6.3.2 Volumetric Pipettes. Class A, assortment. To dilute samples to calibration range of the ion chromatograph (IC).
6.3.3 Ion Chromatograph (IC). Suppressed or non-suppressed, with a conductivity detector and electronic integrator operating in the peak area mode. Other detectors, a strip chart recorder, and peak heights may be used.
7.0 Reagents and Standards.
NOTE: Unless otherwise indicated, all reagents must conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society (ACS reagent grade). When such specifications are not available, the best available grade shall be used.
7.1 Sampling.
7.1.1 filter. Teflon mat (e.g., Pallflex TX40HI45) filter. When the stack gas temperature exceeds 210C (410F) a quartz fiber filter may be used.
7.1.2 Water. Deionized, distilled water that conforms to American Society of Testing and Materials (ASTM) Specification D 1193-77 or 91, Type 3 (incorporated by reference - see 60.17).
7.1.3 Acidic Absorbing Solution, 0.1 N Sulfuric Acid (H2SO4). To prepare 1 L, slowly add 2.80 ml of concentrated 17.9 M H2SO4 to about 900 ml of water while stirring, and adjust the final volume to 1 L using additional water. Shake well to mix the solution.
7.1.4 Silica Gel, Crushed Ice, and Stopcock Grease. Same as Method 5, Sections 7.1.2, 7.1.4, and 7.1.5, respectively.
7.1.5 Alkaline Absorbing Solution, 0.1 N Sodium Hydroxide (NaOH). To prepare 1 L, dissolve 4.00 g of solid NaOH in about 900 ml of water and adjust the final volume to 1 L using additional water. Shake well to mix the solution.
7.1.6 Sodium Thiosulfate, (Na2S2O33.5 H2O).
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in Section 7.1.2.
7.2.2 Absorbing Solution Blanks. A separate blank solution of each absorbing reagent should be prepared for analysis with the field samples. Dilute 200 ml of each absorbing solution (250 ml of the acidic absorbing solution, if a condensate impinger is used) to the same final volume as the field samples using the blank sample of rinse water. If a particulate determination is conducted, collect a blank sample of acetone.
7.2.3 Halide Salt Stock Standard Solutions. Prepare concentrated stock solutions from reagent grade sodium chloride (NaCl), sodium bromide (NaBr), and sodium fluoride (NaF). Each must be dried at 110C (230F) for two or more hours and then cooled to room temperature in a desiccator immediately before weighing. Accurately weigh 1.6 to 1.7 g of the dried NaCl to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact Cl- concentration using Equation 26A-1 in Section 12.2. In a similar manner, accurately weigh and solubilize 1.2 to 1.3 g of dried NaBr and 2.2 to 2.3 g of NaF to make 1-liter solutions. Use Equations 26A-2 and 26A-3 in Section 12.2, to calculate the Br- and F- concentrations. Alternately, solutions containing a nominal certified concentration of 1000 mg/L NaCl are commercially available as convenient stock solutions from which standards can be made by appropriate volumetric dilution. Refrigerate the stock standard solutions and store no longer than one month.
7.2.4 Chromatographic Eluent. Same as Method 26, Section 7.2.4.
7.2.5 Water. Same as Section 7.1.1.
7.2.6 Acetone. Same as Method 5, Section 7.2.
7.3 Quality Assurance Audit Samples.
When making compliance determinations, and upon availability, audit samples may be obtained from the appropriate EPA regional Office or from the responsible enforcement authority.
NOTE: The responsible enforcement authority should be notified at least 30 days prior to the test date to allow sufficient time for sample delivery.
8.0 Sample Collection, Preservation, Storage, and Transport.
NOTE: Because of the complexity of this method, testers and analysts should be trained and experienced with the procedures to ensure reliable results.
8.1 Sampling.
8.1.1 Pretest Preparation.
Follow the general procedure given in Method 5, Section 8.1, except the filter need only be desiccated and weighed if a particulate determination will be conducted.
8.1.2 Preliminary Determinations.
Same as Method 5, Section 8.2.
8.1.3 Preparation of Sampling train.
Follow the general procedure given in Method 5, Section 8.1.3, except for the following variations: Add 50 ml of 0.1 N H2SO4 to the condensate impinger, if used. Place 100 ml of 0.1 N H2SO4 in each of the next two impingers. Place 100 ml of 0.1 N NaOH in each of the following two impingers. Finally, transfer approximately 200-300 g of preweighed silica gel from its container to the last impinger. Set up the train as in Figure 26A-1. When used, the optional cyclone is inserted between the Probe liner and filter holder and located in the heated filter box.
8.1.4 Leak-Check Procedures.
Follow the leak-check procedures given in Method 5, Sections 8.4.2 (Pretest Leak- Check), 8.4.3 (Leak-Checks During the Sample Run), and 8.4.4 (Post-Test Leak-Check).
8.1.5 Sampling train Operation.
Follow the general procedure given in Method 5, Section 8.5. It is important to maintain a temperature around the Probe, filter (and cyclone, if used) of greater than 120C (248 F) since it is extremely difficult to purge acid gases off these components. (These components are not quantitatively recovered and hence any collection of acid gases on these components would result in potential undereporting these emissions. The applicable subparts may specify alternative higher temperatures.) For each run, record the data required on a data sheet such as the one shown in Method 5, Figure 5-3. If the condensate impinger becomes too full, it may be emptied, recharged with 50 ml of 0.1 N H2SO4, and replaced during the sample run. The condensate emptied must be saved and included in the measurement of the volume of moisture collected and included in the sample for analysis. The additional 50 ml of absorbing reagent must also be considered in calculating the moisture. Before the sampling train integrity is compromised by removing the impinger, conduct a leak-check as described in Method 5, Section 8.4.2.
8.1.6 Post-Test Moisture Removal (Optional).
When the optional cyclone is included in the sampling train or when liquid is visible on the filter at the end of a sample run even in the absence of a cyclone, perform the following procedure. Upon completion of the test run, connect the ambient air conditioning tube at the Probe inlet and operate the train with the filter heating system at least 120C (248 F) at a low flow rate (e.g., •H = 1 in. H2O) to vaporize any liquid and hydrogen halides in the cyclone or on the filter and pull them through the train into the impingers. After 30 minutes, turn off the flow, remove the conditioning tube, and examine the cyclone and filter for any visible liquid. If liquid is visible, repeat this step for 15 minutes and observe again. Keep repeating until the cyclone is dry.
NOTE: It is critical that this is repeated until the cyclone is completely dry.
8.2 Sample Recovery.
Allow the Probe to cool. When the Probe can be handled safely, wipe off all the external surfaces of the tip of the Probe probe nozzle and place a cap loosely over the tip to prevent gaining or losing particulate matter. Do not cap the Probe tip tightly while the sampling train is cooling down because this will create a vacuum in the filter holder, drawing water from the impingers into the holder. Before moving the sampling train to the cleanup site, remove the Probe from the sample train, wipe off any silicone grease, and cap the open outlet of the impinger train, being careful not to lose any condensate that might be present. Wipe off any silicone grease and cap the filter or cyclone inlet. Remove the umbilical cord from the last impinger and cap the impinger. If a flexible line is used between the first impinger and the filter holder, disconnect it at the filter holder and let any condensed water drain into the first impinger. Wipe off any silicone grease and cap the filter holder outlet and the impinger inlet. Ground glass stoppers, plastic caps, serum caps, Teflon tape, Parafilm, or aluminum foil may be used to close these openings. Transfer the Probe and filter/impinger assembly to the cleanup area. This area should be clean and protected from the weather to minimize sample contamination or loss. Inspect the train prior to and during disassembly and note any abnormal conditions. Treat samples as follows:
8.2.1 Container No. 1 (Optional; filter Catch for Particulate Determination).
Same as Method 5, Section 8.7.6.1, Container No. 1.
8.2.2 Container No. 2 (Optional; Front-Half Rinse for Particulate Determination).
Same as Method 5, Section 8.7.6.2, Container No. 2.
8.2.3 Container No. 3 (Knockout and Acid impinger Catch for Moisture and Hydrogen Halide Determination).
Disconnect the impingers. Measure the liquid in the acid and knockout impingers to +1 ml by using a graduated cylinder or by weighing it to +0.5 g by using a balance. Record the volume or weight of liquid present. This information is required to calculate the moisture content of the effluent gas. Quantitatively transfer this liquid to a leak-free sample storage container. Rinse these impingers and connecting glassware including the back portion of the filter holder (and flexible tubing, if used) with water and add these rinses to the storage container. Seal the container, shake to mix, and label. The fluid level should be marked so that if any sample is lost during transport, a correction proportional to the lost volume can be applied. Retain rinse water and acidic absorbing solution blanks to be analyzed with the samples.
8.2.4 Container No. 4 (Alkaline impinger Catch for Halogen and Moisture Determination).
Measure and record the liquid in the alkaline impingers as described in Section 8.2.3. Quantitatively transfer this liquid to a leak-free sample storage container. Rinse these two impingers and connecting glassware with water and add these rinses to the container. Add 25 mg of sodium thiosulfate per ppm halogen anticipated to be in the stack gas multiplied by the volume (dscm) of stack gas sampled (0.7 mg/ppm-dscf). Seal the container, shake to mix, and label; mark the fluid level. Retain alkaline absorbing solution blank to be analyzed with the samples.
NOTE: 25 mg per sodium thiosulfate per ppm halogen anticipated to be in the stack includes a safety factor of approximately 5 to assure complete reaction with the hypohalous acid to form a second Cl>- ion in the alkaline solution.
8.2.5 Container No. 5 (Silica Gel for Moisture Determination).
Same as Method 5, Section 8.7.6.3, Container No. 3.
8.2.6 Container Nos. 6 through 9 (Reagent Blanks).
Save portions of the absorbing reagents (0.1 N H2SO4 and 0.1 N NaOH) equivalent to the amount used in the sampling train; dilute to the approximate volume of the corresponding samples using rinse water directly from the wash bottle being used. Add the same ratio of sodium thiosulfate solution used in container No. 4 to the 0.1 N NaOH absorbing reagent blank. Also, save a portion of the rinse water alone and a portion of the acetone equivalent to the amount used to rinse the front half of the sampling train. Place each in a separate, pre-labeled sample container.
8.2.7 Prior to shipment
Prior to shipment, recheck all sample containers to ensure that the caps are well-secured. Seal the lids of all containers around the circumference with Teflon tape. Ship all liquid samples upright and all particulate filters with the particulate catch facing upward.
9.0 Quality Control.
9.1 Miscellaneous Quality Control Measures.
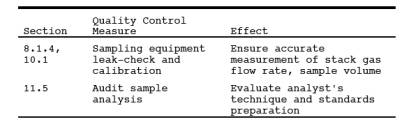
9.1 Volume metering System Checks. Same as Method 5, Section 9.2.
10.0 Calibration and Standardization.
NOTE: Maintain a laboratory log of all calibrations.
10.1 Probe probe nozzle, pitot Tube Assembly, dry gas metering System, Probe Heater, tenperature sensors, Leak-Check of metering System, and barometer.
Same as Method 5, Sections 10.1, 10.2, 10.3, 10.4, 10.5, 8.4.1, and 10.6, respectively.
10.2 Ion Chromatograph.
10.2.1 To prepare the calibration standards, dilute given amounts (1.0 ml or greater) of the stock standard solutions to convenient volumes, using 0.1 N H2SO4 or 0.1 N NaOH, as appropriate. Prepare at least four calibration standards for each absorbing reagent containing the three stock solutions such that they are within the linear range of the field samples.
10.2.2 Using one of the standards in each series, ensure adequate baseline separation for the peaks of interest.
10.2.3 Inject the appropriate series of calibration standards, starting with the lowest concentration standard first both before and after injection of the quality control check sample, reagent blanks, and field samples. This allows compensation for any instrument drift occurring during sample analysis. The values from duplicate injections of these calibration samples should agree within 5 percent of their mean for the analysis to be valid.
10.2.4 Determine the peak areas, or height, of the standards and plot individual values versus halide ion concentrations in g/ml.
10.2.5 Draw a smooth curve through the points. Use linear regression to calculate a formula describing the resulting linear curve.
11.0 Analytical Procedures.
NOTE: the liquid levels in the sample containers and confirm on the analysis sheet whether or not leakage occurred during transport. If a noticeable leakage has occurred, either void the sample or use methods, subject to the approval of the Administrator, to correct the final results.
11.1 Sample Analysis.
11.1.1 The IC conditions will depend upon analytical column type and whether suppressed or non-suppressed IC is used. An example chromatogram from a non-suppressed system using a 150-mm Hamilton PRP-X100 anion column, a 2 ml/min flow rate of a 4 mM 4-hydroxy benzoate solution adjusted to a pH of 8.6 using 1 N NaOH, a 50 l sample loop, and a conductivity detector set on 1.0 S full scale is shown in Figure 26-2.
11.1.2 Before sample analysis, establish a stable baseline. Next, inject a sample of water, and determine if any Cl-, Br-, or F>- appears in the chromatogram. If any of these ions are present, repeat the load/injection procedure until they are no longer present. Analysis of the acid and alkaline absorbing solution samples requires separate standard calibration curves; prepare each according to Section 10.2. Ensure adequate baseline separation of the analyses.
11.1.3 Between injections of the appropriate series of calibration standards, inject in duplicate the reagent blanks, quality control sample, and the field samples. Measure the areas or heights of the Cl-, Br-, and F- peaks. Use the mean response of the duplicate injections to determine the concentrations of the field samples and reagent blanks using the linear calibration curve. The values from duplicate injections should agree within 5 percent of their mean for the analysis to be valid. If the values of duplicate injections are not within 5 percent of the mean, the duplicator injections shall be repeated and all four values used to determine the average response. Dilute any sample and the blank with equal volumes of water if the concentration exceeds that of the highest standard.
11.2 Container Nos. 1 and 2 and Acetone Blank (Optional; Particulate Determination).
Same as Method 5, Sections 11.2.1 and 11.2.2, respectively.
11.3 Container No. 5.
Same as Method 5, Section 11.2.3 for silica gel.
11.4 Audit Sample Analysis.
11.4.1 When the method is used to analyze samples to demonstrate compliance with a source emission regulation, a set of two EPA audit samples must be analyzed, subject to availability.
11.4.2 Concurrently analyze the audit samples and the compliance samples in the same manner to evaluate the technique of the analyst and the standards preparation.
11.4.3 The same analyst, analytical reagents, and analytical system shall be used for the compliance samples and the EPA audit samples. If this condition is met, duplicate auditing of subsequent compliance analyses for the same enforcement agency within a 30-day period is waived. An audit sample set may not be used to validate different sets of compliance samples under the jurisdiction of separate enforcement agencies, unless prior arrangements have been made with both enforcement agencies.
11.5 Audit Sample Results.
11.5.1 Calculate the concentrations in mg/L of audit sample and submit results following the instructions provided with the audit samples.
11.5.2 Report the results of the audit samples and the compliance determination samples along with their identification numbers, and the analyst's name to the responsible enforcement authority. Include this information with reports of any subsequent compliance analyses for the same enforcement authority during the 30-day period.
11.5.3 The concentrations of the audit samples obtained by the analyst shall agree within 10 percent of the actual concentrations. If the 10 percent specification is not met, reanalyze the compliance and audit samples, and include initial and reanalysis values in the test report.
11.5.4 Failure to meet the 10 percent specification may require retests until the audit problems are resolved. However, if the audit results do not affect the compliance or noncompliance status of the affected facility, the Administrator may waive the reanalysis requirement, further audits, or retests and accept the results of the compliance test. While steps are being taken to resolve audit analysis problems, the Administrator may also choose to use the data to determine the compliance or noncompliance status of the affected facility.
12.0. Data Analysis and Calculations.
NOTE: Retain at least one extra decimal figure beyond those contained in the available data in intermediate calculations, and round off only the final answer appropriately.
12.1 Nomenclature. Same as Method 5, Section 12.1. In addition:
BX- = Mass concentration of applicable absorbing solution blank, g halide ion (Cl-, Br>-, F-) /ml, not to exceed 1 g/ml which is 10 times the published analytical detection limit of 0.1 g/ml. (It is also approximately 5 percent of the mass concentration anticipated to result from a one hour sample at 10 ppmv HCl.)
C = Concentration of hydrogen halide (HX) or halogen (X2), dry basis, mg/dscm.
K = 10-3 mg/g.
KHCl = 1.028 (g HCl/g-mole)/(g Cl>-/g-mole).
KHBr = 1.013 (g HBr/g-mole)/(g Br>-/g-mole).
KHF = 1.053 (g HF/g-mole)/(g F>-/g-mole).
mHX = Mass of HCl, HBr, or HF in sample, ug.
mX2 = Mass of Cl2 or Br2 in sample, ug.
SX- = Analysis of sample, ug halide ion (Cl-, Br-,F-)/ml.
Vs = Volume of filtered and diluted sample, ml.
12.2 Calculate the exact Cl-, Br-, and F concentration in the halide salt stock standard solutions using the following equations.

12.3 Average dry gas meter temperature and Average Orifice Pressure Drop. See data sheet (Figure 5-3 of Method 5).
12.4 Dry Gas Volume. Calculate Vm(std) and adjust for leakage, if necessary, using the equation in Section 12.3 of Method 5.
12.5 Volume of Water Vapor and Moisture Content. Calculate the volume of water vapor Vw(std) and moisture content B>ws from the data obtained in this method (Figure 5-3 of Method 5); use Equations 5-2 and 5-3 of Method 5.
12.6 Isokinetic Variation and Acceptable Results. Use Method 5, Section 12.11.
12.7 Acetone Blank Concentration, Acetone Wash Blank Residue Weight, Particulate Weight, and Particulate Concentration. For particulate determination.
12.8 Total g HCl, HBr, or HF Per Sample.
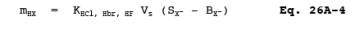
12.9 Total g Cl2 or Br2 Per Sample.

12.10 Concentration of Hydrogen Halide or Halogen in flue Gas.

12.11 Stack Gas Velocity and Volumetric flow Rate. Calculate the average stack gas velocity and volumetric flow rate, if needed, using data obtained in this method and the equations in Sections 12.3 and 12.4 of Method 2.
13.0 Method Performance.
13.1 Precision and Bias.
The method has a possible measurable negative bias below 20 ppm HCl perhaps due to reaction with small amounts of moisture in the Probe and filter. Similar bias for the other hydrogen halides is possible.
13.2 Sample Stability.
The collected Cl- samples can be stored for up to 4 weeks for analysis for HCl and Cl2.
13.3 Detection Limit.
A typical analytical detection limit for HCl is 0.2 g/ml. Detection limits for the other analyses should be similar. Assuming 300 ml of liquid recovered for the acidified impingers and a similar amounts recovered from the basic impingers, and 1 dscm of stack gas sampled, the analytical detection limits in the stack gas would be about 0.04 ppm for HCl and Cl2, respectively.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
1. Steinsberger, S. C. and J. H. Margeson. Laboratory and Field Evaluation of a Methodology for Determination of Hydrogen Chloride Emissions from Municipal and Hazardous Waste Incinerators. U.S. Environmental Protection Agency, Office of Research and Development. Publication No. 600/3-89/064. April 1989. Available from National Technical Information Service, Springfield, VA 22161 as PB89220586/AS.
2. State of California Air Resources Board. Method 421 - Determination of Hydrochloric Acid Emissions from Stationary Sources. March 18, 1987.
3. Cheney, J.L. and C.R. Fortune. Improvements in the Methodology for Measuring Hydrochloric Acid in Combustion Source Emissions. J. Environ. Sci. Health. A19(3): 337-350. 1984.
4. Stern, D.A., B.M. Myatt, J.F. Lachowski, and K.T. McGregor. Speciation of Halogen and Hydrogen Halide Compounds in Gaseous Emissions. In: Incineration and Treatment of Hazardous Waste: Proceedings of the 9th Annual Research Symposium, Cincinnati, Ohio, May 2-4, 1983. Publication No. 600/9-84-015. July 1984. Available from National Technical Information Service, Springfield, VA 22161 as PB84-234525.
5. Holm, R.D. and S.A. Barksdale. Analysis of Anions in Combustion Products. In: Ion Chromatographic Analysis of Environmental Pollutants, E. Sawicki, J.D. Mulik, and E. Wittgenstein (eds.). Ann Arbor, Michigan, Ann Arbor Science Publishers. 1978. pp. 99-110.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
Figure 26A-1. Sampling train
Warning: include(right-column/Services.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 337
Warning: include(): Failed opening 'right-column/Services.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 337
Warning: include(right-column/Products.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 338
Warning: include(): Failed opening 'right-column/Products.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 338
Warning: include(right-column/Resources.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 339
Warning: include(): Failed opening 'right-column/Resources.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 339
Warning: include(right-column/Experts.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 340
Warning: include(): Failed opening 'right-column/Experts.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 340
Warning: include(common-epa/right_col_method_ads.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 343
Warning: include(): Failed opening 'common-epa/right_col_method_ads.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 343
Warning: include(right-column/UniqueValue.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 344
Warning: include(): Failed opening 'right-column/UniqueValue.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 344
Warning: include(right-column/QualityCheck.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 345
Warning: include(): Failed opening 'right-column/QualityCheck.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 345
Warning: include(common/allstats.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 352
Warning: include(): Failed opening 'common/allstats.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-26a.php on line 352