Warning: include(common/header.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 22
Warning: include(): Failed opening 'common/header.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 22
Warning: include(common-home/leftcolumn_epa.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 30
Warning: include(): Failed opening 'common-home/leftcolumn_epa.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 30
EPA Methods List with Links
US EPA Method 7d - Determination Of Nitrogen Oxide Emissions From Stationary Sources (Alkaline-Permanganate/Ion Chromatographic Method)
NOTE: This method is not inclusive with respect to specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) essential to its performance. Some material is incorporated by reference from other methods in this part. Therefore, to obtain reliable results, persons using this method should have a thorough knowledge of at least the following additional test methods: Method 1, Method 3, Method 6, Method 7, and Method 7C.
1.0 Scope and Application.
1.1 Analytes.
| Analyte | CAS No. | Sensitivity |
| Nitrogen oxides (NOx), as NO2, including: Nitric oxide (NO) Nitrogen dioxide (NO2) |
10102-43-9 10102-44-0 |
7 ppmv |
1.2 Applicability.
This method applies to the measurement of NOx emissions from fossil-fuel fired steam generators, electric utility plants, nitric acid plants, or other sources as specified in the regulations.
1.3 Data Quality Objectives.
Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods.
2.0 Summary of Method.
An integrated gas sample is extracted from the stack and passed through impingers containing an alkaline- potassium permanganate solution; NOx (NO + NO2) emissions are oxidized to NO3-. Then NO3- is analyzed by ion chromatography.
3.0 Definitions. [Reserved]
4.0 Interferences.
Same as in Method 7C, Section 4.0.
5.0 Safety.
5.1 Disclaimer.
This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and to determine the applicability of regulatory limitations prior to performing this test method.
5.2 Corrosive reagents.
The following reagents are hazardous. Personal protective equipment and safe procedures are useful in preventing chemical splashes. If contact occurs, immediately flush with copious amounts of water for at least 15 minutes. Remove clothing under shower and decontaminate. Treat residual chemical burns as thermal burns.
5.2.1 Hydrogen Peroxide (H2O2).
Irritating to eyes, skin, nose, and lungs. 30% H2O2 is a strong oxidizing agent; avoid contact with skin, eyes, and combustible material. Wear gloves when handling.
5.2.2 Sodium Hydroxide (NaOH).
Causes severe damage to eye tissues and to skin. Inhalation causes irritation to nose, throat, and lungs. Reacts exothermically with limited amounts of water.
5.2.3 Potassium Permanganate (KMnO4).
Caustic, strong oxidizer. Avoid bodily contact with.
6.0 Equipment and Supplies.
6.1 Sample Collection and Sample Recovery.
Same as Method 7C, Section 6.1. A schematic of the sampling train used in performing this method is shown in Figure 7C-1 of Method 7C.
6.2 Sample Preparation and Analysis.
6.2.1 Magnetic Stirrer.
With 25- by 10-mm Teflon- coated stirring bars.
6.2.2 filtering Flask.
500-ml capacity with sidearm.
6.2.3 Buchner Funnel.
75-mm ID, with spout equipped with a 13-mm ID by 90-mm long piece of Teflon tubing to minimize possibility of aspirating sample solution during filtration.
6.2.4 filter Paper.
Whatman GF/C, 7.0-cm diameter.
6.2.5 Stirring Rods.
6.2.6 Volumetric Flask. 250-ml.
6.2.7 Pipettes. Class A.
6.2.8 Erlenmeyer Flasks. 250-ml.
6.2.9 Ion Chromatograph.
Equipped with an anion separator column to separate NO3-, H+ suppressor, and necessary auxiliary equipment. Nonsuppressed and other forms of ion chromatography may also be used provided that adequate resolution of NO3- is obtained. The system must also be able to resolve and detect NO2-.
7.0 Reagents and Standards.
NOTE: Unless otherwise indicated, it is intended that all reagents conform to the specifications established by the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available; otherwise, use the best available grade.
7.1 Sample Collection.
7.1.1 Water. Deionized distilled to conform to ASTM specification D 1193-77 or 91 Type 3 (incorporated by reference - see 60.17).
7.1.2 Potassium Permanganate, 4.0 Percent (w/w), Sodium Hydroxide, 2.0 Percent (w/w). Dissolve 40.0 g of KMnO4 and 20.0 g of NaOH in 940 ml of water.
7.2 Sample Preparation and Analysis.
7.2.1 Water. Same as in Section 7.1.1.
7.2.2 Hydrogen Peroxide (H2O2), 5 Percent. Dilute 30 percent H2O2 1:5 (v/v) with water.
7.2.3 Blank Solution. Dissolve 2.4 g of KMnO4 and 1.2 g of NaOH in 96 ml of water. Alternatively, dilute 60 ml of KMnO4/NaOH solution to 100 ml.
7.2.4 KNO3 Standard Solution. Dry KNO3 at 110EC for 2 hours, and cool in a desiccator. Accurately weigh 9 to 10 g of KNO3 to within 0.1 mg, dissolve in water, and dilute to 1 liter. Calculate the exact NO3- concentration using Equation 7D-1 in Section 12.2. This solution is stable for 2 months without preservative under laboratory conditions.
7.2.5 Eluent, 0.003 M NaHCO3/0.0024 M Na2CO3. Dissolve 1.008 g NaHCO3 and 1.018 g Na2CO3 in water, and dilute to 4 liters. Other eluents capable of resolving nitrate ion from sulfate and other species present may be used.
7.2.6 Quality Assurance Audit Samples. Same as Method 7, Section 7.3.10. When requesting audit samples, specify that they be in the appropriate concentration range for Method 7D.
8.0 Sample Collection, Preservation, Transport, and Storage.
8.1 Sampling.
Same as in Method 7C, Section 8.1.
8.2 Sample Recovery.
Same as in Method 7C, Section 8.2.
8.3 Sample Preparation for Analysis.
NOTE: Samples must be analyzed within 28 days of collection.
8.3.1 Note the level of liquid in the sample container, and determine whether any sample was lost during shipment. If a noticeable amount of leakage has occurred, the volume lost can be determined from the difference between initial and final solution levels, and this value can then be used to correct the analytical result. Quantitatively transfer the contents to a 1-liter volumetric flask, and dilute to volume.
8.3.2 Sample preparation can be started 36 hours after collection. This time is necessary to ensure that all NO2- is converted to NO3- in the collection solution. Take a 50-ml aliquot of the sample and blank, and transfer to 250- ml Erlenmeyer flasks. Add a magnetic stirring bar. Adjust the stirring rate to as fast a rate as possible without loss of solution. Add 5 percent H2O2 in increments of approximately 5 ml using a 5-ml pipette. When the KMnO4 color appears to have been removed, allow the precipitate to settle, and examine the supernatant liquid. If the liquid is clear, the H2O2 addition is complete. If the KMnO4 color persists, add more H2O2, with stirring, until the supernatant liquid is clear.
NOTE: The faster the stirring rate, the less volume of H2O2 that will be required to remove the KMnO4.) Quantitatively transfer the mixture to a Buchner funnel containing GF/C filter paper, and filter the precipitate. The spout of the Buchner funnel should be equipped with a 13-mm ID by 90-mm long piece of Teflon tubing. This modification minimizes the possibility of aspirating sample solution during filtration. filter the mixture into a 500- ml filtering flask. Wash the solid material four times with water. When filtration is complete, wash the Teflon tubing, quantitatively transfer the filtrate to a 250-ml volumetric flask, and dilute to volume. The sample and blank are now ready for NO3- analysis.
9.0 Quality Control.
| Section | Quality Control Measure | Effect |
| 8.2, 10.1-10.3 | Sampling equipment leak-check and calibration | Ensure accurate measurement of sample volume |
| 10.4 | Spectrophotometer calibration | Ensure linearity of spectrophotometer response to standards |
| 11.3 | Spiked sample analysis | Ensure reduction efficiency of column |
| 11.6 | Audit sample analysis | Evaluate analytical technique, preparation of standards |
10.0 Calibration and Standardizations.
10.1 dry gas meter (DGM) System.
10.1.1 Initial calibration.
Same as in Method 6, Section 10.1.1. For detailed instructions on carrying out this calibration, it is suggested that Section 3.5.2 of Citation 4 in Section 16.0 of Method 7C be consulted.
10.1.2 Post-Test calibration Check.
Same as in Method 6, Section 10.1.2.
10.2 Thermometers for DGM and barometer.
Same as in Method 6, Sections 10.2 and 10.4, respectively.
10.3 Ion Chromatograph.
10.3.1 Dilute a given volume (1.0 ml or greater) of the KNO3 standard solution to a convenient volume with water, and use this solution to prepare calibration standards. Prepare at least four standards to cover the range of the samples being analyzed. Use pipettes for all additions. Run standards as instructed in Section 11.2. Determine peak height or area, and plot the individual values versus concentration in μg NO3-/ml.
10.3.2 Do not force the curve through zero. Draw a smooth curve through the points. The curve should be linear. With the linear curve, use linear regression to determine the calibration equation.
11.0 Analytical Procedures.
11.1 The following chromatographic conditions are recommended: 0.003 M NaHCO3/0.0024 Na2CO3 eluent solution (Section 7.2.5), full scale range, 3 μMHO; sample loop, 0.5 ml; flow rate, 2.5 ml/min. These conditions should give a NO3- retention time of approximately 15 minutes (Figure 7D-1).
11.2 Establish a stable baseline. Inject a sample of water, and determine whether any NO3- appears in the chromatogram. If NO3- is present, repeat the water load/injection procedure approximately five times; then re- inject a water sample and observe the chromatogram. When no NO3- is present, the instrument is ready for use. Inject calibration standards. Then inject samples and a blank. Repeat the injection of the calibration standards (to compensate for any drift in response of the instrument). Measure the NO3- peak height or peak area, and determine the sample concentration from the calibration curve.
11.3 Audit Analysis. Same as in Method 7, Section 11.4.
12.0 Data Analysis and Calculations.
Carry out calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Nomenclature.
Same as in Method 7C, Section 12.1.
12.2 NO3- concentration.
Calculate the NO3- concentration in the KNO3 standard solution (see Section 7.2.4) using the following equation:
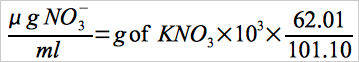 Eq. 7D-1
Eq. 7D-112.3 Sample Volume, Dry Basis, Corrected to Standard Conditions.
Same as in Method 7C, Section 12.4.
12.4 Total μg NO2 Per Sample.
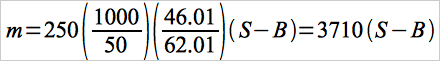 Eq. 7D-2
Eq. 7D-2where:
| 250 | = | Volume of prepared sample, ml. |
| 1000 | = | Total volume of KMnO4 solution, ml. |
| 50 | = | Aliquot of KMnO4/NaOH solution, ml. |
| 46.01 | = | Molecular weight of NO2-. |
| 62.01 | = | Molecular weight of NO3-. |
12.5 Sample Concentration.
Same as in Method 7C, Section 12.7.
13.0 Method Performance.
13.1 Precision.
The intra-laboratory relative standard deviation for a single measurement is approximately 6 percent at 200 to 270 ppm NOx.
13.2 Bias.
The method does not exhibit any bias relative to Method 7.
13.3 Range.
The lower detectable limit is similar to that of Method 7C. No upper limit has been established; however, when using the recommended sampling conditions, the method has been found to collect NOx emissions quantitatively up to 1782 mg NOx/m3, as NO2 (932 ppm NOx).
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 References.
Same as Method 7C, Section 16.0, References 1, 2, 4, and 5.
17.0 Tables, Diagrams, flowcharts, and Validation Data.
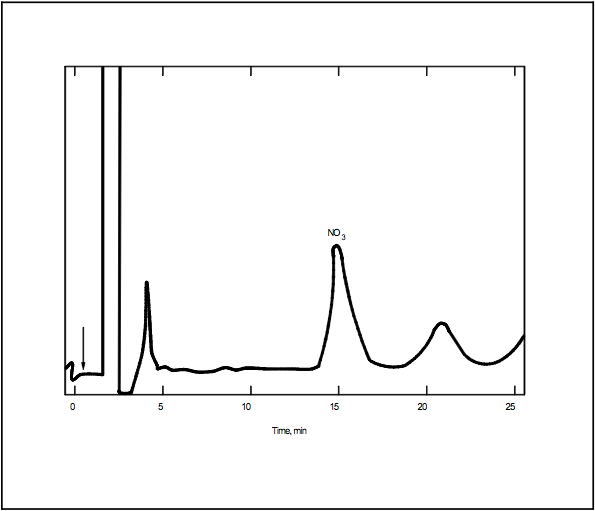
Figure 7D-1. Ion Chromatograph of a Prepared Sample.
Warning: include(right-column/Services.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 240
Warning: include(): Failed opening 'right-column/Services.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 240
Warning: include(right-column/Products.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 241
Warning: include(): Failed opening 'right-column/Products.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 241
Warning: include(right-column/Resources.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 242
Warning: include(): Failed opening 'right-column/Resources.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 242
Warning: include(right-column/Experts.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 243
Warning: include(): Failed opening 'right-column/Experts.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 243
Warning: include(common-epa/right_col_method_ads.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 246
Warning: include(): Failed opening 'common-epa/right_col_method_ads.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 246
Warning: include(right-column/UniqueValue.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 247
Warning: include(): Failed opening 'right-column/UniqueValue.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 247
Warning: include(right-column/QualityCheck.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 248
Warning: include(): Failed opening 'right-column/QualityCheck.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 248
Warning: include(common/allstats.php): Failed to open stream: No such file or directory in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 255
Warning: include(): Failed opening 'common/allstats.php' for inclusion (include_path='.:/opt/homebrew/Cellar/php/8.3.4/share/php/pear') in /Users/lowerlevel/Sites/D7036/epamethods/air-test-methods/m-07d.php on line 255